Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
La regolazione dei livelli di colesterolo ematico dipende principalmente da due organi:
L’INTESTINO: assorbe colesterolo dal cibo o dalla bile IL FEGATO: produce colesterolo che viene utilizzato dagli organi periferici (o eliminato con la bile)
")
3
H COLESTEROLO

5
Extrinsic (dietary) cholesterol and fatty acid transport
Extrinsic (dietary) cholesterol and fatty acid transport. Dietary cholesterol and fatty acids are absorbed by enterocytes in the duodenum and proximal jejunum. These lipids are then esterified and packed into chylomicrons in association with the apolipoproteins apoB48 and apoAI. After entering the circulation through the thoracic duct, chylomicrons acquire the apolipoproteins apoE, apoCI, apoCII, and apoCIII from HDL. Circulating chylomicrons are depleted of triglycerides by the action of lipoprotein lipase, in a reaction that is dependent on apoCII. After lipoprotein lipase has removed a large proportion of the triglyceride core, chylomicrons lose many of their apolipoproteins; the resulting lipoprotein is termed a chylomicron remnant. The liver takes up these remnants in an interactions mediated by apoE binding to the LDL receptor or to the LDL-related receptor (not shown)
 cholesterol and fatty acid transport. Dietary cholesterol and fatty acids are absorbed by enterocytes in the duodenum and proximal jejunum. These lipids are then esterified and packed into chylomicrons in association with the apolipoproteins apoB48 and apoAI. After entering the circulation through the thoracic duct, chylomicrons acquire the apolipoproteins apoE, apoCI, apoCII, and apoCIII from HDL. Circulating chylomicrons are depleted of triglycerides by the action of lipoprotein lipase, in a reaction that is dependent on apoCII. After lipoprotein lipase has removed a large proportion of the triglyceride core, chylomicrons lose many of their apolipoproteins; the resulting lipoprotein is termed a chylomicron remnant. The liver takes up these remnants in an interactions mediated by apoE binding to the LDL receptor or to the LDL-related receptor (not shown)")
6
Endogenous cholesterol and fatty acid transport
Endogenous cholesterol and fatty acid transport. The liver produces nascent VLDL particles by combining triglyceride with apoplipoprotein apoB100 (the required structural protein) and other small apolipoproteins, including apoE, apoCI, apoCII, apoCIII and apoAII; this reaction is dependent on microsomal triglyceride transfer protein (MTP). Nascent VLDL particles become mature VLDL as circulating HDL enriches the VLDL with additional apolipoproteins. The triglyceride core of VLDL is removed by the action of lipoprotein lipase on the endothelial cells of adipose and muscle tissue. Depletion of the triglyceride core results in the formation of VLDL remnants (IDL and LDL). IDL can be removed from the circulation via LDL receptor (LDL-R)-mediated uptake or hepatic lipase can hydrolyze the remaining triglycerides, resulting in the formation of LDL. LDL is removed from the circulation by LDL-R-mediated uptake by the liver or other tissues. Alternatively, LDL can be oxidized and taken up by macrophages, in a reaction that depends on the scavenger receptor-A (SR-A); this reaction results in the formation of foam cells. Note the many points of intersection between HDL and endogenous lipid metabolism.
 and other small apolipoproteins, including apoE, apoCI, apoCII, apoCIII and apoAII; this reaction is dependent on microsomal triglyceride transfer protein (MTP). Nascent VLDL particles become mature VLDL as circulating HDL enriches the VLDL with additional apolipoproteins. The triglyceride core of VLDL is removed by the action of lipoprotein lipase on the endothelial cells of adipose and muscle tissue. Depletion of the triglyceride core results in the formation of VLDL remnants (IDL and LDL). IDL can be removed from the circulation via LDL receptor (LDL-R)-mediated uptake or hepatic lipase can hydrolyze the remaining triglycerides, resulting in the formation of LDL. LDL is removed from the circulation by LDL-R-mediated uptake by the liver or other tissues. Alternatively, LDL can be oxidized and taken up by macrophages, in a reaction that depends on the scavenger receptor-A (SR-A); this reaction results in the formation of foam cells. Note the many points of intersection between HDL and endogenous lipid metabolism.")
7
HDL metabolism. HDL originates in the liver or the intestine or from remnant lipoprotein products released during the hydrolysis of lipoproteins by plasma liporotein lipase. Nascent HDL is cholesterol poor; it contains mainly phospholipid and the apolipoprotein apoAI. Nascent HDL circulates in the plasma and receives free cholesterol from cholesterol laden cells,including macrophages, by a process that is depndent on the enzyme ATP-binding cassette transporter A! (ABCA1). Using apoAI as a cofactor, plasma lecithin:cholesterol acyl transferase (LCAT) esterifies the free cholesterol, resulting in alrger hydrophobic core of cholesteryl esters (small HDL). HDL becomes larger as it accumulates more cholestery esters. HDL can transfer cholesteryl esters to VLDL and IDL via the enzyme cholesteryl ester transfer protein (CETP) and accumulate triglyceride from the VLDL and IDL. The realtive triglycerdie rich HDL can then be eliminated by one of three mechanisms. First HDL can be taken up directly by the hepatic SR-B1 receptor. Second, hepatic lipase can hydrolyze the triglyceride core, regenerating small HDL. Third, HDL can acquire the apolipoprotein apoE; this particle can then be taken up by the hepatic LDL-R
. Using apoAI as a cofactor, plasma lecithin:cholesterol acyl transferase (LCAT) esterifies the free cholesterol, resulting in alrger hydrophobic core of cholesteryl esters (small HDL). HDL becomes larger as it accumulates more cholestery esters. HDL can transfer cholesteryl esters to VLDL and IDL via the enzyme cholesteryl ester transfer protein (CETP) and accumulate triglyceride from the VLDL and IDL. The realtive triglycerdie rich HDL can then be eliminated by one of three mechanisms. First HDL can be taken up directly by the hepatic SR-B1 receptor. Second, hepatic lipase can hydrolyze the triglyceride core, regenerating small HDL. Third, HDL can acquire the apolipoprotein apoE; this particle can then be taken up by the hepatic LDL-R.")
8
Disfunzione endoteliale produzione di NO e PGI2 Espressione
molecole di adesione (VCAM1, ICAM, selectina) Reclutamento monociti macrofagi Imaging targets in atherothrombosis. Illustration of processes of atherogenesis ranging from pre-lesional endothelial dysfunction (left) through monocyte recruitment to the development of advanced plaque complicated by thrombosis (right). The mechanisms are grossly simplified but focus on components (for example, cell adhesion molecules, macrophages, connective tissue elements, lipid core and fibrin) and processes (for example, apoptosis, proteolysis, angiogenesis and thrombosis) in plaques that have been imaged or that present useful potential imaging targets. Symbols indicate the feasibility (+ or -) of imaging using each of the modalities listed (see text for details). ICAM, intercellular cell adhesion molecule; LDL, low-density lipoprotein; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; NO, nitric oxide; PET, positron emission tomography; VCAM, vascular cell adhesion molecule.
 Reclutamento. monociti. macrofagi. Imaging targets in atherothrombosis. Illustration of processes of atherogenesis ranging from pre-lesional endothelial dysfunction (left) through monocyte recruitment to the development of advanced plaque complicated by thrombosis (right). The mechanisms are grossly simplified but focus on components (for example, cell adhesion molecules, macrophages, connective tissue elements, lipid core and fibrin) and processes (for example, apoptosis, proteolysis, angiogenesis and thrombosis) in plaques that have been imaged or that present useful potential imaging targets. Symbols indicate the feasibility (+ or -) of imaging using each of the modalities listed (see text for details). ICAM, intercellular cell adhesion molecule; LDL, low-density lipoprotein; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; NO, nitric oxide; PET, positron emission tomography; VCAM, vascular cell adhesion molecule.")
9
LDL and atherosclerosis
LDL and atherosclerosis. Elevated LDL is a major risk factor for the development of atherosclerosis. Native LDL that migrates into the subendothelial space can undergo chemical transformation tyo oxidized LDL via lipid peroxidation and fragmentation of apoB100. Oxidized LDL has a number of deleterious effects on vascular function. Oxidized LDL promotes monocyte chemotaxis into the subendothelial space (A) and inhibits monocyte egress from that space (B). Resident monocyte-macrophages bind to oxidized LDL via a scavenger receptor (SR-A), resulting in the formation of lipid-laden foam cells (C). Oxidized LDL can directly injure endothelial cells and cause endothelial dysfunction (D). Oxidized LDL can also cause foam cell necrosis, with release of numerous proteolyitic enzymes that can damage the intima (E)
 and inhibits monocyte egress from that space (B). Resident monocyte-macrophages bind to oxidized LDL via a scavenger receptor (SR-A), resulting in the formation of lipid-laden foam cells (C). Oxidized LDL can directly injure endothelial cells and cause endothelial dysfunction (D). Oxidized LDL can also cause foam cell necrosis, with release of numerous proteolyitic enzymes that can damage the intima (E)")
10
Development of vascular-wall lesions
Development of vascular-wall lesions. a | Schematic of the early sub-endothelial accumulation of monocytes, macrophages and foam cells, and the formation of fatty streaks. b | At the site of a nascent vascular lesion, lipids, lipoproteins or other reactive substances stimulate the endothelium to produce inflammatory cytokines, chemoattractants and other reactive molecules. The cytokine-activated endothelium expresses adhesion molecules that lead to the recruitment of peripheral blood monocytes to the inflammatory site. Low-affinity interactions between monocytes and the endothelium, which are mediated by selectins and integrins, lead to capture and rolling of monocytes on the endothelial surface. On the basis of studies in genetically modified mice, E- and P-selectins have been implicated in the development of vascular lesions. On activation of monocytes by endothelial cell products such as chemokines, monocyte integrins achieve high-affinity interactions with endothelial adhesion molecules, and cells arrest on the endothelial surface. Several mouse studies have implicated the 4 1-integrin (also known as VLA-4) and its cognate ligand VCAM-1 in these high-affinity interactions. Activated macrophages within the lesion secrete chemotactic products, including chemokines. In response to these chemokine gradients, cells migrate through the endothelium. Many lines of evidence implicate the chemokine receptor CCR2 in this process, but preliminary human genetic data indicate that CCR5 and CX3CR1 might also have a role. c | As cells transmigrate across the endothelium, they are activated by the milieu of inflammatory cytokines that are secreted by the inflamed endothelium and the underlying smooth muscle layer. They differentiate into the metabolically active, secretory and highly phagocytic inflammatory macrophage. As macrophages accumulate, they take up lipoproteins and actively accumulate lipid to become foam cells. CCR2, chemokine (CC) motif receptor 2; CCR5, chemokine (CC) motif receptor 5; CX3CR1, chemokine (CX3C) motif receptor 1; VCAM-1, vascular cell-adhesion molecule 1; VLA-4, very late (activation) antigen 4.
 and its cognate ligand VCAM-1 in these high-affinity interactions. Activated macrophages within the lesion secrete chemotactic products, including chemokines. In response to these chemokine gradients, cells migrate through the endothelium. Many lines of evidence implicate the chemokine receptor CCR2 in this process, but preliminary human genetic data indicate that CCR5 and CX3CR1 might also have a role. c | As cells transmigrate across the endothelium, they are activated by the milieu of inflammatory cytokines that are secreted by the inflamed endothelium and the underlying smooth muscle layer. They differentiate into the metabolically active, secretory and highly phagocytic inflammatory macrophage. As macrophages accumulate, they take up lipoproteins and actively accumulate lipid to become foam cells. CCR2, chemokine (CC) motif receptor 2; CCR5, chemokine (CC) motif receptor 5; CX3CR1, chemokine (CX3C) motif receptor 1; VCAM-1, vascular cell-adhesion molecule 1; VLA-4, very late (activation) antigen 4.")
11
The macrophage/foam cell and its role in lipid metabolism
The macrophage/foam cell and its role in lipid metabolism. Macrophage/foam cells are important in the metabolism of lipids and lipoprotein particles within the vascular wall. Modified low-density lipoprotein (mod-LDL) is endocytosed into the macrophage/foam cell after binding to the macrophage scavenger receptor MSR-A. The endocytosed particles are transported to the lysosomes, and free cholesterol (FC) is then released into the cytosol. Cytosolic FC is kept in appropriate equilibrium with cholesterol ester (CE) through the action of two enzymes: acyl-coenzyme A cholesterol-acetyltransferase (ACAT) and neutral cholesterol-ester hydrolases (NCEH). Activation of the macrophage/foam cell nuclear receptor LXR by FC induces the synthesis of the cholesterol transporter ABCA1 and the important lipoprotein APOE, thereby stimulating cholesterol efflux from the cell. ABCA1, ATP-binding cassette, subfamily A, member 1; APOE, apolipoprotein E; LXR, nuclear oxysterol receptor.
 is endocytosed into the macrophage/foam cell after binding to the macrophage scavenger receptor MSR-A. The endocytosed particles are transported to the lysosomes, and free cholesterol (FC) is then released into the cytosol. Cytosolic FC is kept in appropriate equilibrium with cholesterol ester (CE) through the action of two enzymes: acyl-coenzyme A cholesterol-acetyltransferase (ACAT) and neutral cholesterol-ester hydrolases (NCEH). Activation of the macrophage/foam cell nuclear receptor LXR by FC induces the synthesis of the cholesterol transporter ABCA1 and the important lipoprotein APOE, thereby stimulating cholesterol efflux from the cell. ABCA1, ATP-binding cassette, subfamily A, member 1; APOE, apolipoprotein E; LXR, nuclear oxysterol receptor.")
12
Secrezione Proteasi Produzione ROS apoptosi Produzione
citochine e GF Proliferazione m. liscia deposizione connettivo Aggregazione piastrinica Trombosi Imaging targets in atherothrombosis. Illustration of processes of atherogenesis ranging from pre-lesional endothelial dysfunction (left) through monocyte recruitment to the development of advanced plaque complicated by thrombosis (right). The mechanisms are grossly simplified but focus on components (for example, cell adhesion molecules, macrophages, connective tissue elements, lipid core and fibrin) and processes (for example, apoptosis, proteolysis, angiogenesis and thrombosis) in plaques that have been imaged or that present useful potential imaging targets. Symbols indicate the feasibility (+ or -) of imaging using each of the modalities listed (see text for details). ICAM, intercellular cell adhesion molecule; LDL, low-density lipoprotein; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; NO, nitric oxide; PET, positron emission tomography; VCAM, vascular cell adhesion molecule.
 through monocyte recruitment to the development of advanced plaque complicated by thrombosis (right). The mechanisms are grossly simplified but focus on components (for example, cell adhesion molecules, macrophages, connective tissue elements, lipid core and fibrin) and processes (for example, apoptosis, proteolysis, angiogenesis and thrombosis) in plaques that have been imaged or that present useful potential imaging targets. Symbols indicate the feasibility (+ or -) of imaging using each of the modalities listed (see text for details). ICAM, intercellular cell adhesion molecule; LDL, low-density lipoprotein; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; NO, nitric oxide; PET, positron emission tomography; VCAM, vascular cell adhesion molecule.")
13
Overview of high-density lipoprotein metabolism and potential targets for therapeutic intervention. Synthesis of new high-density lipoprotein (HDL) particles begins with the secretion of apolipoprotein A-I (apoA-I) from the liver. Fibrates have been shown to increase the expression of apoA-I in human hepatocytes. Lipid-free or lipid-poor apoA-I can subsequently serve as an acceptor for ABC transporter A1 (ABCA1)-mediated lipid efflux from hepatocytes or macrophages. Infusion of apoA-I has been shown to attenuate atherosclerosis in animals and possibly in humans. ABCA1-mediated lipid efflux from macrophages can also be enhanced by transcriptional upregulation of this lipid transporter through the nuclear receptors liver X receptor (LXR)/retinoid X receptor (RXR)18 or retinoic acid receptor- (RAR )128. ABCA1-mediated efflux of cholesterol and phospholipids results in the formation of pre- or nascent HDL particles that are further modified by lecithin-cholesterol acyltransferase (LCAT). The resulting HDL2 (larger, less dense particles) and HDL3 (smaller, more dense particles) can serve as acceptors for ABCG1-mediated cholesterol efflux. Expression of this transporter can also be stimulated by LXR activation. Infusion of recombinant phospholipid–apoA-I complexes and large unilamellar phospholipid vesicles (LUVs) have been shown to increase HDL levels, presumably by acting as acceptors for ABCG1-mediated cholesterol efflux. Cholesterol esters from HDL can be transferred to apoB-containing lipoproteins by the action of cholesteryl ester transfer protein (CETP). Inhibition of CETP in humans has recently been shown to increase HDL and lower low-density lipoprotein (LDL) cholesterol. The catabolism of HDL can also be inhibited by nicotinic acid through a mechanism that is largely unknown. Finally, HDL cholesterol can be taken up by the liver and subsequently secreted into the bile in a process that is mediated by scavenger receptor BI (SR-BI).
 particles begins with the secretion of apolipoprotein A-I (apoA-I) from the liver. Fibrates have been shown to increase the expression of apoA-I in human hepatocytes. Lipid-free or lipid-poor apoA-I can subsequently serve as an acceptor for ABC transporter A1 (ABCA1)-mediated lipid efflux from hepatocytes or macrophages. Infusion of apoA-I has been shown to attenuate atherosclerosis in animals and possibly in humans. ABCA1-mediated lipid efflux from macrophages can also be enhanced by transcriptional upregulation of this lipid transporter through the nuclear receptors liver X receptor (LXR)/retinoid X receptor (RXR)18 or retinoic acid receptor- (RAR )128. ABCA1-mediated efflux of cholesterol and phospholipids results in the formation of pre- or nascent HDL particles that are further modified by lecithin-cholesterol acyltransferase (LCAT). The resulting HDL2 (larger, less dense particles) and HDL3 (smaller, more dense particles) can serve as acceptors for ABCG1-mediated cholesterol efflux. Expression of this transporter can also be stimulated by LXR activation. Infusion of recombinant phospholipid–apoA-I complexes and large unilamellar phospholipid vesicles (LUVs) have been shown to increase HDL levels, presumably by acting as acceptors for ABCG1-mediated cholesterol efflux. Cholesterol esters from HDL can be transferred to apoB-containing lipoproteins by the action of cholesteryl ester transfer protein (CETP). Inhibition of CETP in humans has recently been shown to increase HDL and lower low-density lipoprotein (LDL) cholesterol. The catabolism of HDL can also be inhibited by nicotinic acid through a mechanism that is largely unknown. Finally, HDL cholesterol can be taken up by the liver and subsequently secreted into the bile in a process that is mediated by scavenger receptor BI (SR-BI)..")
14
Mechanisms of cholesterol efflux from the arterial wall
Mechanisms of cholesterol efflux from the arterial wall. On entering the sub-endothelial space, lipid-free or lipid-poor apolipoprotein A-I (apoA-I) can bind to the ABC transporter A1 (ABCA1) on the cell surface of macrophages in the arterial wall and promote efflux of free cholesterol and phospholipids from these cells. This results in the formation of nascent high-density lipoprotein (HDL) particles, which undergo further modification by the lecithin-cholesterol acyltransferase (LCAT) enzyme and develop into spherically shaped HDL2 (larger, less dense particles) or HDL3 (smaller, more dense particles), which, in turn, can act as acceptors for ABCG1-mediated cholesterol efflux from macrophages, resulting in further cholesterol enrichment of HDL, before returning to the circulation. Although inter-conversion of HDL subspecies is depicted as occurring in the arterial wall, it probably also occurs in the plasma.
 can bind to the ABC transporter A1 (ABCA1) on the cell surface of macrophages in the arterial wall and promote efflux of free cholesterol and phospholipids from these cells. This results in the formation of nascent high-density lipoprotein (HDL) particles, which undergo further modification by the lecithin-cholesterol acyltransferase (LCAT) enzyme and develop into spherically shaped HDL2 (larger, less dense particles) or HDL3 (smaller, more dense particles), which, in turn, can act as acceptors for ABCG1-mediated cholesterol efflux from macrophages, resulting in further cholesterol enrichment of HDL, before returning to the circulation. Although inter-conversion of HDL subspecies is depicted as occurring in the arterial wall, it probably also occurs in the plasma.")
15
Pleiotropic effects of HDL in the vessel wall
Pleiotropic effects of HDL in the vessel wall. Several pleiotropic effects of HDL in the vasculature may underlie its anti-atherogenicity. These include: (1) inhibition of chemotaxis of monocytes; (2) inhibition of monocyte adhesion to endothelial cells; (3) inhibition of LDL oxidation; (4) inhibition of OX-LDL-induced endothelial dysfunction and apoptosis; (5) stimulation of endothelial cell proliferation; (6) stimulation of endothelial synthesis of prostacyclin and CNP; (7) stimulation of cholesterol efflux from macrophages and foam cells; (8) stimulation of smooth muscle cell (SMC) proliferation; (9) inhibition of platelet activation; (10) inhibition of factor X activation and stimulation of activated protein C.
 inhibition of chemotaxis of monocytes; (2) inhibition of monocyte adhesion to endothelial cells; (3) inhibition of LDL oxidation; (4) inhibition of OX-LDL-induced endothelial dysfunction and apoptosis; (5) stimulation of endothelial cell proliferation; (6) stimulation of endothelial synthesis of prostacyclin and CNP; (7) stimulation of cholesterol efflux from macrophages and foam cells; (8) stimulation of smooth muscle cell (SMC) proliferation; (9) inhibition of platelet activation; (10) inhibition of factor X activation and stimulation of activated protein C.")
16
FATTORI CHE DETERMINANO LIVELLI BASSI DI HDL COLESTEROLO
Eccessiva assunzione di carboidrati (>60% dell’apporto calorico totale) Stile di vita sedentario Obesità/sovrappeso Sindrome metabolica Diabete mellito di tipo II Ipertrigliceridemie
 Stile di vita sedentario. Obesità/sovrappeso. Sindrome metabolica. Diabete mellito di tipo II. Ipertrigliceridemie.")
17
Hypertriglyceridemia and HDL metabolism: hypothesized mechanisms
Hypertriglyceridemia and HDL metabolism: hypothesized mechanisms. In hypertriglyceridemic states, there is an increased mass transfer of triglycerides (TG) from TG-rich lipoproteins to HDL particles through the action of cholesteryl ester transfer protein (CETP), a process leading to TG enrichment of HDL. TG-enriched HDL have been shown to be more prone to lipolysis by hepatic lipase (HL), giving rise to the formation of small, lipolytically modified HDL particles. Apolipoprotein (apo) A-I may be shed from the particle in this process. There are also data to suggest that apo A-I may be in a more dissociable form on TG-enriched HDL, possibly due to a change in the particle stability. These mechanisms may all be responsible to a significant extent for the increased fractional catabolic rate (FCR) of apo A-I generally seen in hypertriglyceridemic states and ultimately, for the concomitant reductions in plasma HDL cholesterol levels. HDL metabolism in hypertriglyceridemic states: an overview, Clinica Chimica Acta, Volume 286, Issues 1-2, August 1999, Pages Benoit Lamarche, Shirya Rashid and Gary F. Lewis
 from TG-rich lipoproteins to HDL particles through the action of cholesteryl ester transfer protein (CETP), a process leading to TG enrichment of HDL. TG-enriched HDL have been shown to be more prone to lipolysis by hepatic lipase (HL), giving rise to the formation of small, lipolytically modified HDL particles. Apolipoprotein (apo) A-I may be shed from the particle in this process. There are also data to suggest that apo A-I may be in a more dissociable form on TG-enriched HDL, possibly due to a change in the particle stability. These mechanisms may all be responsible to a significant extent for the increased fractional catabolic rate (FCR) of apo A-I generally seen in hypertriglyceridemic states and ultimately, for the concomitant reductions in plasma HDL cholesterol levels. HDL metabolism in hypertriglyceridemic states: an overview, Clinica Chimica Acta, Volume 286, Issues 1-2, August 1999, Pages Benoit Lamarche, Shirya Rashid and Gary F. Lewis.")
18
FATTORI CHE DETERMINANO LIVELLI BASSI DI HDL COLESTEROLO
Eccessiva assunzione di carboidrati (>60% dell’apporto calorico totale) Stile di vita sedentario Obesità/sovrappeso Sindrome metabolica Diabete mellito di tipo II Ipertrigliceridemie Ipo--lipoproteinemia primaria Insufficienza renale cronica Sindrome nefrotica AIDS/lipodistrofia Uso di alcuni farmaci (diuretici tiazidici, β-bloccanti, steroidi anabolizzanti, progestinici, agenti antiretrovirali) Fumo
 Stile di vita sedentario. Obesità/sovrappeso. Sindrome metabolica. Diabete mellito di tipo II. Ipertrigliceridemie. Ipo--lipoproteinemia primaria. Insufficienza renale cronica. Sindrome nefrotica. AIDS/lipodistrofia. Uso di alcuni farmaci (diuretici tiazidici, β-bloccanti, steroidi anabolizzanti, progestinici, agenti antiretrovirali) Fumo.")
19
Cellular regulation of cholesterol metabolism
Cellular regulation of cholesterol metabolism. Binding of apolipoprotein B100to LDL receptors (LDL-R)promotes LDL internalization intoendocytic vesicles and fusion of the vesicles with lysosomes. LDL-R is recycled to the cell surface, whilethe lipoprotein particle is hydrolyzed into aminoacids and free cholestero. Intracellular cholesterol has three regulatory effects on the cell. First, cholesterol decreases the activity of HGM CoA reductase, the rate-limiting enzyme in cholesterol synthesis. Second, cholesterol activates acetyl CoA:cholesterol acyltransferase (ACAT), an enzyme that esterifies free cholesterol into cholesteryl ester (cholesteryl oleate) that can be stored in the cell. Third, cholesterol inhibits the transcription of the gene encoding the LDL receptor, and thereby decreases further uptake of cholesterol by the cell.
promotes LDL internalization intoendocytic vesicles and fusion of the vesicles with lysosomes. LDL-R is recycled to the cell surface, whilethe lipoprotein particle is hydrolyzed into aminoacids and free cholestero. Intracellular cholesterol has three regulatory effects on the cell. First, cholesterol decreases the activity of HGM CoA reductase, the rate-limiting enzyme in cholesterol synthesis. Second, cholesterol activates acetyl CoA:cholesterol acyltransferase (ACAT), an enzyme that esterifies free cholesterol into cholesteryl ester (cholesteryl oleate) that can be stored in the cell. Third, cholesterol inhibits the transcription of the gene encoding the LDL receptor, and thereby decreases further uptake of cholesterol by the cell.")
21
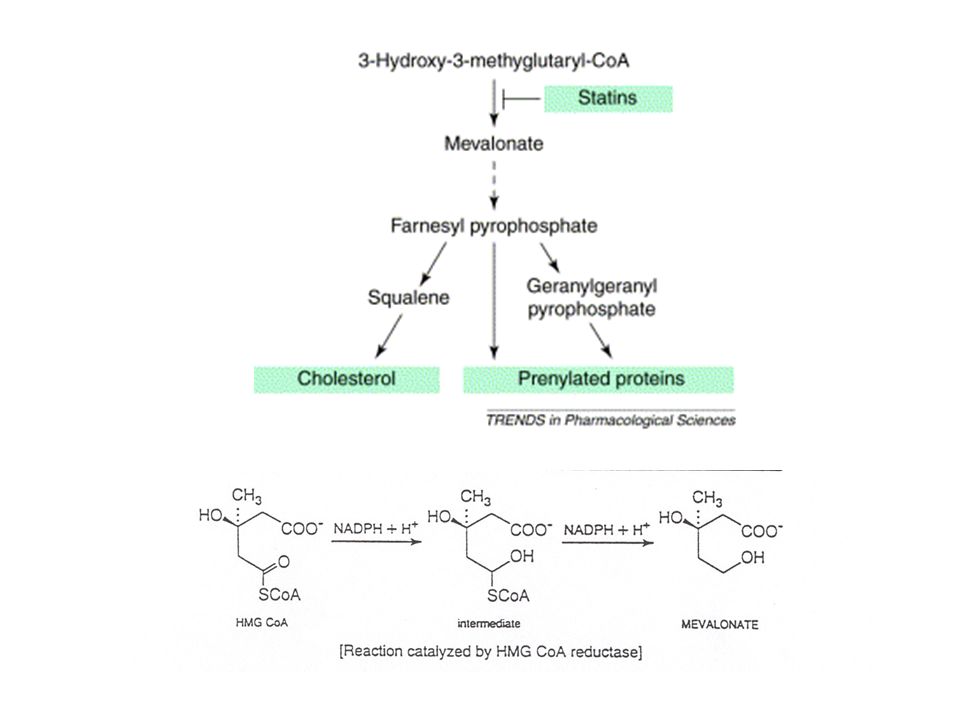
STATINE: INIBITORI DELLA HMGCoA REDUTTASI

22
LDL-R expression Effects of statins on cholesterol metabolism. Statins competitively inhibit HGM CoA reductase, the enzyme that catalyzes a crucial step in cholesterol synthesis. Decreased hepatocyte cholesterol concentration leads to protease activation and cleavage of the sterol regulatory element binding protein (SREBP), which is a transcription factor that normally resides in the cytoplasm. The cleaved SREBP diffuses into the nucleus, where it binds to sterol response elements (SRE), leading to up-regulation of LDL-receptor (LDL-R) gene transcription. This leads to increased hepatocyte LDL-R expression, which mediates increased plasma LDL clearance and results in reduced circulating LDL levels.
, which is a transcription factor that normally resides in the cytoplasm. The cleaved SREBP diffuses into the nucleus, where it binds to sterol response elements (SRE), leading to up-regulation of LDL-receptor (LDL-R) gene transcription. This leads to increased hepatocyte LDL-R expression, which mediates increased plasma LDL clearance and results in reduced circulating LDL levels.")
23
EFFETTI DELLE STATINE CHE NON DIPENDONO DALL’AZIONE SUI LIVELLI DI LDL
stabilizzazione del mRNA per la eNOS infiltrazione dei monociti nella parete arteriosa produzione di metalloproteinasi da parte dei macrofagi proliferazione delle cellule muscolari lisce e apoptosi suscettibilità delle LDL alla ossidazione aggregazione piastrinica azione anti-infiammatoria

24
FIBRATI

25
Effects of fibrates on lipid metabolism
Effects of fibrates on lipid metabolism. Fibrates have several effects on lipid metabolism, all of whihc are thought to result from PPARalpha-mediated changes in gene transcription. PPARalpha activation also decreases hepatic synthesis of apoCIII, which normally inhibits lipoprotein lipase activity. The decrease apoCIII, combined with incerased lipoprotein lipase expression in muscle vascular beds, leads to increased fatty acid uptake in muscle cells and increased fatty acid oxidation. PPARalpha also increases fatty acid oxidation in hepatocytes. The end result of these metabolic alterations is a decrease in plasma triglyceride levels and an increase in plasma HDL levels. LOD levels also decrease modestly because of a decrease in hepatic fatty acid and triglyceride synthesis (not shown)
")
26
Fig. 3. Heterodimerization of peroxisome proliferator activated receptor (PPAR- ) with the retinoid X receptor (RXR) produces an active transcription complex that binds to a peroxisome-proliferator-response element (PPRE). In the absence of ligand, the heterodimer forms high-affinity complexes with nuclear co-repressor proteins, such as nuclear receptor co-repressor (N-CoR), which prevent transcriptional activation by sequestration of the receptor complex from the promoter. Dissociation of co-repressors occurs as a consequence of a ligand-induced conformational change, and the activated heterodimer can then bind to the PPRE. This results in activation or suppression of transcription of a target gene. Recently, co-activators such as PPAR- co-activator 1 (PGC-1) have been identified, which promote the assembly of an effective transcriptional complex that includes histone acetyltransferases (HATs) and steroid receptor co-activator-1 (SR-1). Abbreviation: cis-RA, cis-retinoic acid.
 with the retinoid X receptor (RXR) produces an active transcription complex that binds to a peroxisome-proliferator-response element (PPRE). In the absence of ligand, the heterodimer forms high-affinity complexes with nuclear co-repressor proteins, such as nuclear receptor co-repressor (N-CoR), which prevent transcriptional activation by sequestration of the receptor complex from the promoter. Dissociation of co-repressors occurs as a consequence of a ligand-induced conformational change, and the activated heterodimer can then bind to the PPRE. This results in activation or suppression of transcription of a target gene. Recently, co-activators such as PPAR- co-activator 1 (PGC-1) have been identified, which promote the assembly of an effective transcriptional complex that includes histone acetyltransferases (HATs) and steroid receptor co-activator-1 (SR-1). Abbreviation: cis-RA, cis-retinoic acid..")
27
Effects of fibrates on lipid metabolism
Effects of fibrates on lipid metabolism. Fibrates have several effects on lipid metabolism, all of whihc are thought to result from PPARalpha-mediated changes in gene transcription. PPARalpha activation also decreases hepatic synthesis of apoCIII, which normally inhibits lipoprotein lipase activity. The decrease apoCIII, combined with incerased lipoprotein lipase expression in muscle vascular beds, leads to increased fatty acid uptake in muscle cells and increased fatty acid oxidation. PPARalpha also increases fatty acid oxidation in hepatocytes. The end result of these metabolic alterations is a decrease in plasma triglyceride levels and an increase in plasma HDL levels. LOD levels also decrease modestly because of a decrease in hepatic fatty acid and triglyceride synthesis (not shown)
")
28
EFFETTI DEI FIBRATI Stimolazione della lipoproteina lipasi
produzione epatica di VLDL uptake epatico di LDL Attivazione del recettore PPAR (peroxisome proliferator-activated receptor ) fibrinogeno plasmatico inibizione dell’espressione di NFB
 fibrinogeno plasmatico. inibizione dell’espressione di NFB.")
29
NIACINA o ACIDO NICOTINICO
AZIONI: Blocco della lipolisi dei TG da parte della lipasi ormone-sensibile nel tessuto adiposo Aumento dell’attività della lipoproteina lipasi Diminuzione clearance frazionale di apoAI

30
Effects of niacin on lipid metabolism
Effects of niacin on lipid metabolism. Niacin lowers trigyceride and LDL levels while increasing HDL. The molecular mechanismo of niacin action is unknown, but niacin has been shown to decrease hormone-sensitive lipase activity in adipose tissue, leading to decreased free fatty acid flux to the liver. This decreased free fatty acid flux results in decrease epatic triglyceride synthesis and decrease VLD synthesis. Because LDL is derived from VLDL, the decreased VLDL synthesis decreases plasma concentration of LDL. Niacin also increases the half-life of apoAI, an important apolipoprotein in HDL the increased apoAI levels directly increases levels of plasma HDL, and may also augment reverse cholesterol transport, delivery of cholesterol from HDL to the liver and excretion opf cholesterol in the bile.

31
RESINE A SCAMBIO IONICO
Colestyramine Legano gli acidi biliari e impediscono l’assorbimento del colesterolo intestinale impedendone la solubilizzazione in micelle

33
NUOVI INTERVENTI IN CORSO DI SVILUPPO CLINICO
Inibizione dell’assorbimento intestinale di colesterolo Agisce sulla proteina Niemann-Pick C1-like 1 (NPC1L1) che media l’assorbimento del colesterolo da parte di enterociti e epatociti
 che media l’assorbimento del colesterolo da parte di enterociti e epatociti.")
34
NUOVI INTERVENTI IN CORSO DI SVILUPPO CLINICO
Inibizione dell’assorbimento intestinale di colesterolo Inibizione dell’attività della proteina CETP

35
NUOVI INTERVENTI IN CORSO DI SVILUPPO CLINICO
Inibizione dell’assorbimento intestinale di colesterolo Inibizione dell’attività della proteina CETP Attivazione dei fattori nucleari PPAR/

36
NUOVI INTERVENTI IN CORSO DI SVILUPPO CLINICO
Inibizione dell’assorbimento intestinale di colesterolo Inibizione dell’attività della proteina CETP Attivazione dei fattori nucleari PPAR/ Attivazione del fattore nucleare LXR

37
NUOVI INTERVENTI IN CORSO DI SVILUPPO CLINICO
Inibizione dell’assorbimento intestinale di colesterolo Inibizione dell’attività della proteina CETP Attivazione dei fattori nucleari PPAR/ Attivazione del fattore nucleare LXR Infusione con complessi Apo A-I Milano/fosfolipide

38
OLIO DI PESCE Contiene acidi grassi 3 (eicosapentaenoici, docosaexaenoico) TG plasmatici ( colesterolo)
 .")
Presentazioni simili











 STEATOSI CIRROSI (8-25%) HCC.>")




>")



