Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
EPILESSIA Un male antico, già presente nel trattato di medicina babilonese compilato tra gli anni a.C. dove è designato col nome di miqtu (malattia che fa cadere) e dove le sue principali espressioni cliniche sono accuratamente descritte
 e dove le sue principali espressioni cliniche sono accuratamente descritte.")
2
L’epilessia è una condizione caratterizzata dal ripetersi di crisi epilettiche, definibili come eventi clinici accessuali legati a scariche eccessive, sincrone e abnormi di gruppi di neuroni a livello della corteccia cerebrale. E’ ormai accettato che il termine si debba riferire all’occorrenza di almeno 2 crisi. Inoltre per epilessia, oggi, non s’intende più una singola malattia, ma un insieme di condizioni estremamente diverse tra loro sia sul piano dei fattori che la determinano, sia su quello clinico e prognostico.

3
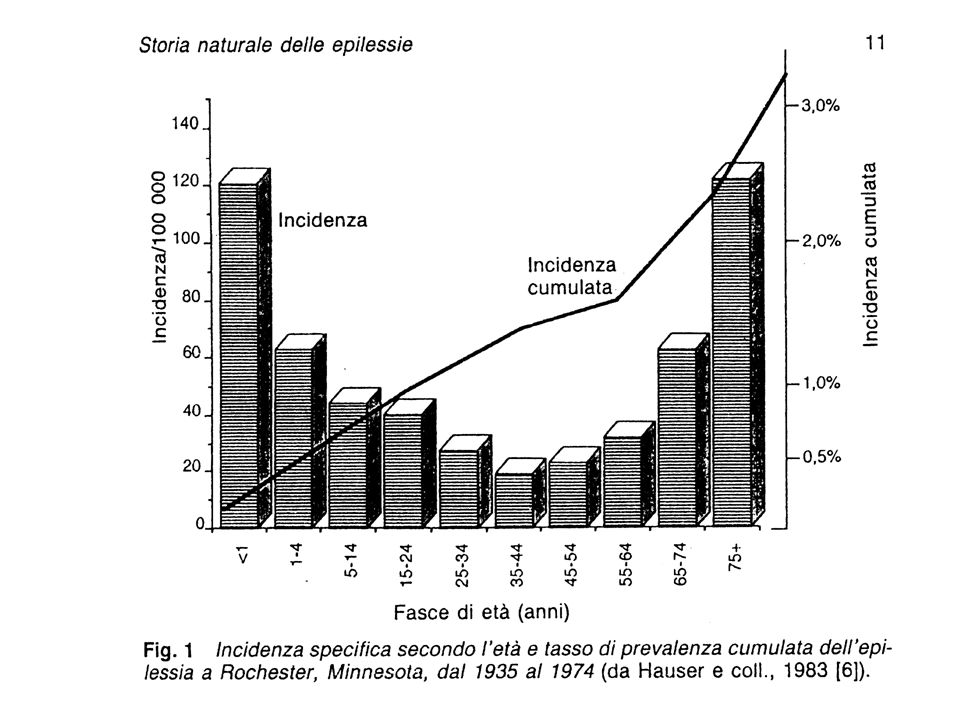
L’epilessia è una patologia frequente che solo in Italia interessa oltre 400 mila persone e fa registrare ogni anno da 20 mila a 40 mila nuovi casi. Si manifesta soprattutto in età infantile o avanzata, ma può colpire qualunque periodo della vita. Negli Stati Uniti i costi sociali di questa malattia sono al terzo posto dopo quelli per le patologie cardio-vascolari e da deficit intellettivo e sensoriale

5
Nell’epilessia si distinguono forme cliniche diverse per eziologia, presentazione clinica e prognosi. Se la scarica neuronale epilettogena inizia e si mantiene localizzata ad una popolazione neuronale ristretta, le crisi sono definite parziali ed hanno fenomenologia coerente con le funzioni delle specifiche aree corticali interessate. Se la scarica inizia localmente, ma si diffonde più o meno rapidamente a vaste aree corticali, vi può essere una generalizzazione secondaria (con manifestazioni convulsive).
.")
6
Le crisi primitivamente generalizzate sono invece sostenute da una scarica epilettica che fin dall’esordio interessa contemporaneamente ampie aree corticali di ambedue gli emisferi. Molte delle crisi descritte come generalizzate, di tipo convulsivo, sono crisi parziali generate da una scarica locale che si diffonde rapidamente senza permettere l’identificazione di fenomeni parziali iniziali.

7
EZIOLOGIA fattori genetici patologia organica riconoscibile (ad esempio, gli esiti lesionali di un trauma cranico, di una infezione, di un ictus) causa rimane sconosciuta. Clinicamente, le crisi epilettiche possono avere natura e manifestazioni diverse e si inseriscono in contesti neurologici estremamente variabili. Nelle epilessie sintomatiche, la disfunzione epilettogena è determinata da alterazioni indotte dalla lesione sulle cellule nervose del tessuto circostante (come nei tumori) o incluse nel tessuto patologico (come nelle displasie).
 o incluse nel tessuto patologico (come nelle displasie).")
8
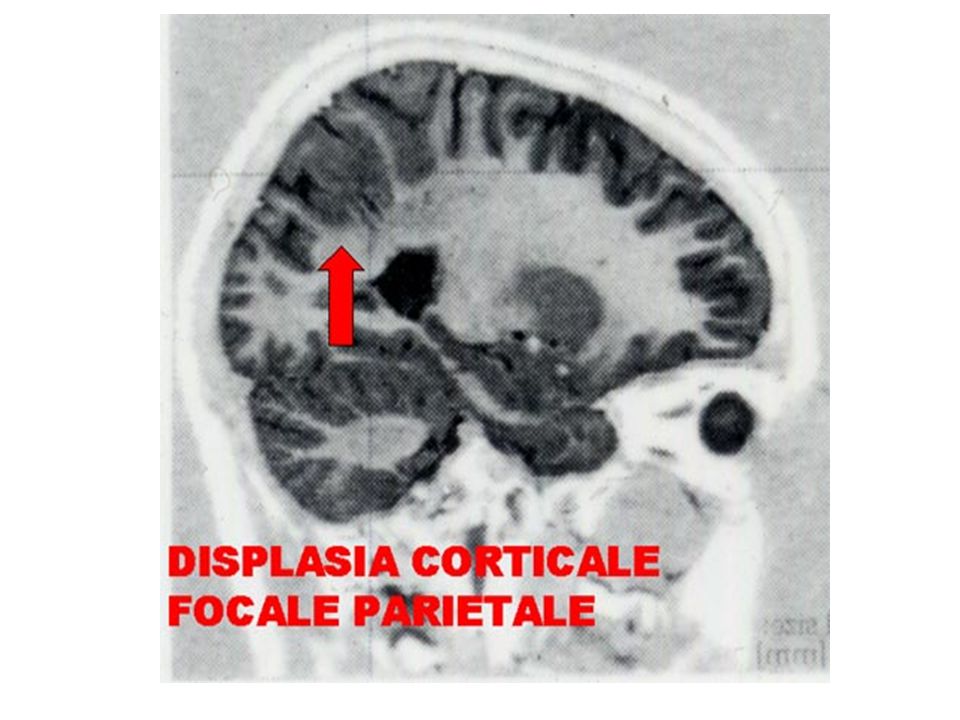
EZIOLOGIA Le crisi possono verificarsi per disfunzione di popolazioni neuronali, in assenza di alterazioni morfologiche (epilessie idiopatiche). Le crisi possono essere secondarie ad un danno cerebrale recente o preesistente, accertato (epilessie sintomatiche) o presunto (epilessie criptogenetiche) L’evoluzione della diagnostica neuroradiologica (risonanza magnetica) ha permesso di identificare un danno cerebrale misconosciuto in un notevole numero di casi: ad es. l’identificazione di aspetti malformativi localizzati, come le displasie corticali focali
 o presunto (epilessie criptogenetiche) L’evoluzione della diagnostica neuroradiologica (risonanza magnetica) ha permesso di identificare un danno cerebrale misconosciuto in un notevole numero di casi: ad es. l’identificazione di aspetti malformativi localizzati, come le displasie corticali focali.")
9
EZIOLOGIA Tra le cause più importanti di epilessia sintomatica sono le malformazioni (displasie, lissencefalia, facomatosi, malformazioni vascolari), le encefalopatie fetali e perinatali su base anossica ed emorragica, le encefalopatie infettive e postraumatiche, le vasculopatie (vasculiti, incidenti embolici), i tumori primitivi o secondari del sistema nervoso, le cromosomopatie (trisomia 18, sindrome di Down di Angelmann e di Prader-Willy) le encefalopatie progressive geneticamente determinate (mioclono-epilessie oltre alle encefalopatie mitocondriali, le acidurie organiche, le aminoacidopati e le malattie perossisomial)i
, le encefalopatie fetali e perinatali su base anossica ed emorragica, le encefalopatie infettive e postraumatiche, le vasculopatie (vasculiti, incidenti embolici), i tumori primitivi o secondari del sistema nervoso, le cromosomopatie (trisomia 18, sindrome di Down di Angelmann e di Prader-Willy) le encefalopatie progressive geneticamente determinate (mioclono-epilessie oltre alle encefalopatie mitocondriali, le acidurie organiche, le aminoacidopati e le malattie perossisomial)i.")
10
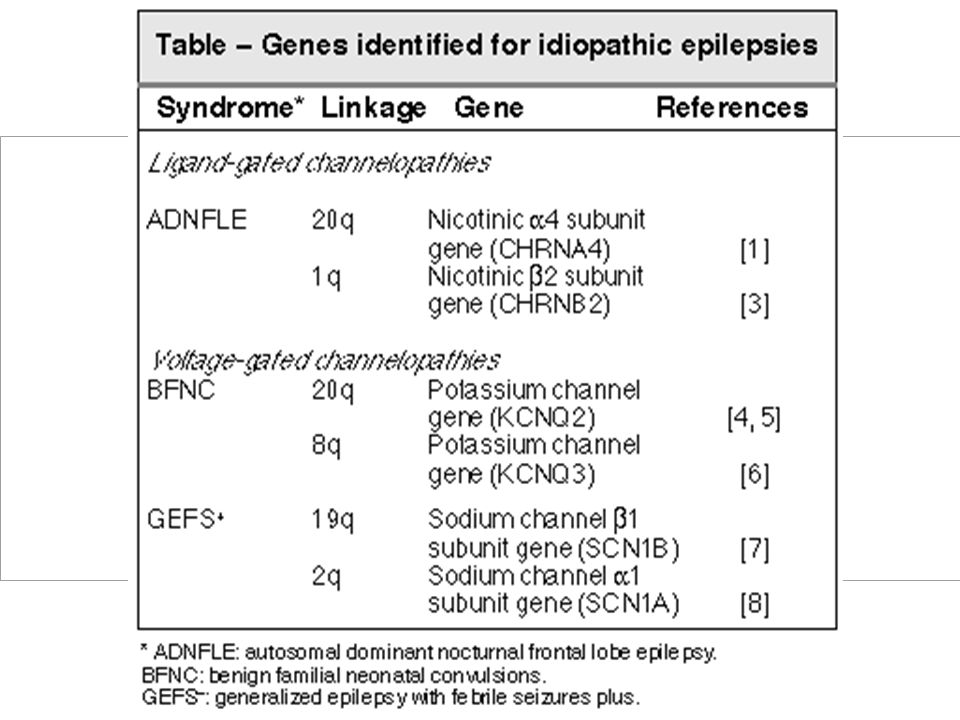
EZIOLOGIA Nelle E. idiopatiche non esiste alcun danno cerebrale dimostrabile: una origine genetica è dimostrata o altamente probabile. Le mutazioni responsabili sono state individuate solo in forme rare a trasmissione dominante, che si esprimono con convulsioni ripetute in epoca neonatale o infantile precoce, con convulsioni febbrili o con crisi parziali che persistono in età adulta.

11
Sono di grande interesse in quanto riguardano geni che codificano per subunità di canali ionici voltaggio-dipendenti o associati a recettori. Le mutazioni identificate interessano il recettore muscarinico in forme famigliari di epilessia frontale, i canali K+ nelle crisi benigne neonatali famigliari, i canali Na+ in una forma particolare di epilessia generalizzata con crisi febbrili, il recettore GABA in una sua variante e in una sotto forma di epilessia mioclonica giovanile. Per le forme più comuni di epilessia idiopatica non si è però giunti alla caratterizzazione molecolare.

13
EZIOLOGIA Dati sperimentali suggeriscono, in epilessie sintomatiche e criptogenetiche, la possibilità che l’attività epilettica possa di per sé indurre alterazioni del tessuto cerebrale interessato con progressiva evoluzione peggiorativa dell’area epilettogena fino all’instaurarsi di refrattarietà alla terapia. Esempio tipico della potenzialità evolutiva del processo epilettogeno è l’epilessia del lobo temporale mesiale. L’età del paziente e la sua storia clinica dovranno sempre orientare le indagini diagnostiche. Danni cerebrali statici epilettogeni prenatali o perinatali si manifestano con crisi ad esordio precoce, Molte encefalopatie “progressive” iniziano in età ben delimitata (infantile o giovanile). Al contrario, alcune lesioni cerebrali acquisite (quali le neoplasie) sono più spesso identificabili nell’adulto.
. Al contrario, alcune lesioni cerebrali acquisite (quali le neoplasie) sono più spesso identificabili nell’adulto.")
15
Per la classificazione delle crisi e della specifica forma di epilessia hanno valore fondamentale, oltre alla clinica, le alterazioni elettroencefalografiche (EEG) concomitanti (EEG critico), molto diverse nelle differenti tipologie di crisi
 concomitanti (EEG critico), molto diverse nelle differenti tipologie di crisi")
19
La correlazione tra fenomenologia clinica/EEG può essere analizzata in registrazione combinata video-EEG. L’inquadramento diagnostico di una specifica forma di epilessia si basa non solo sul tipo di crisi presentate dal soggetto ma anche sulle caratteristiche dell’EEG intercritico, sull’età di esordio della sintomatologia, sulla presenza-assenza di segni clinici/radiologici di danno del SNCe, sulla presenza-assenza di fattori causali identificabili, sulla famigliarità. Sulla base di questi elementi si formula una diagnosi sindromica di forma di epilessia, presupposto fondamentale per programmare ulteriori accertamenti mirati a definirne l’eziologia e a impostare la terapia. La tabella riporta una versione semplificata della classificazione delle sindromi epilettiche elaborata dalla International League Against Epilepsy.

21
Per quanto riguarda la prognosi, vi sono forme
Benigne, che guariscono spontaneamente, Forme responsive alla terapia farmacologia Forme farmaco-resistenti (circa un terzo del totale) Alcune forme resistenti ai farmaci possono rispondere bene al trattamento chirurgico. L’epilessia, a causa del ripetersi di crisi in momenti non prevedibili, può provocare limitazioni di vario genere, difficoltà di inserimento scolastico e lavorativo, e problematiche di ordine psicologico e sociale.
 Alcune forme resistenti ai farmaci possono rispondere bene al trattamento chirurgico. L’epilessia, a causa del ripetersi di crisi in momenti non prevedibili, può provocare limitazioni di vario genere, difficoltà di inserimento scolastico e lavorativo, e problematiche di ordine psicologico e sociale.")
22
Ipereccitabilita’ neuronale: tendenza di un
Fisiopatologia Ipereccitabilita’ neuronale: tendenza di un aggregato neuronale a scaricare in modo eccessive e sincrono Una attivazione neuronale eccessiva dipende da uno stato di ipereccitabilità che si esprime nella tendenza a generare scariche ripetute in risposta a stimoli che normalmente dovrebbero evocare un singolo potenziale d'azione.

23
Fisiopatologia Una attivazione ipersincrona indica la
capacità di un aggregato di neuroni di generare in maniera sincrona una serie di potenziali d'azione: l’applicazione di uno stimolo elettrico o di sostanze convulsivanti ad un’area corticale determina una depolarizzazione seguita da scariche sincrone continue, successivamente diffuse, che costituiscono la crisi

24
Fisiopatologia Meccanismi elettrofisiologici:
i neuroni corticali, per poter generare una scarica epilettica, sviluppino modificazioni transitorie delle proprietà di membrana. Un fenomeno di particolare significato epilettogeno è il cosiddetto paroxysmal depolarizing shift (PDS): brusca depolarizzazione di membrana a cui si associa una scarica di potenziali d'azione multipli. Il PDS si caratterizza, per la lunga durata, l'elevata frequenza di scarica, e la tendenza a manifestarsi in modo sincrono su aggregati neuronali più o meno estesi.
: brusca depolarizzazione di membrana a cui si associa una scarica di potenziali d azione multipli. Il PDS si caratterizza, per la lunga durata, l elevata frequenza di scarica, e la tendenza a manifestarsi in modo sincrono su aggregati neuronali più o meno estesi.")
25
FISIOPATOLOGIA Lo studio di modelli sperimentali di epilessia ha evidenziato la potenzialità epilettogena di alterazioni dei meccanismi di eccitabilità neuronale, che dipendono dai flussi di correnti ioniche attraverso i canali della membrana cellulare. Un enorme avanzamento delle conoscenze è derivato dalla recente definizione della struttura molecolare dei canali e dei geni che codificano le proteine che li costituiscono

26
Meccanismi che regolano le proprietà di membrana ? 1
Pompe ioniche: nella regolazione della omeostasi cellulare è riconosciuto il ruolo di meccanismi di pompa che regolano i flussi nei compartimenti intra- ed extracellulare di Na/K (fuoriuscita del Na, ed ingresso del K), e del Ca (fuoriuscita dal compartimento extracellulare). Tali pompe si basano su trasporti ATP-dipendenti. Una compromissione di tali meccanismi di trasporto causa un aumento del Na e del Ca intracellulare, e conseguente depolarizzazione di membrana. Può verificarsi in condizioni di ridotto apporto energetico ai neuroni corticali, ad esempio durante ipoglicemia od anossia, facilitando l'innesco di scariche di tipo epilettico
, e del Ca (fuoriuscita dal compartimento extracellulare). Tali pompe si basano su trasporti ATP-dipendenti. Una compromissione di tali meccanismi di trasporto causa un aumento del Na e del Ca intracellulare, e conseguente depolarizzazione di membrana. Può verificarsi in condizioni di ridotto apporto energetico ai neuroni corticali, ad esempio durante ipoglicemia od anossia, facilitando l innesco di scariche di tipo epilettico.")
27
Meccanismi che regolano le proprietà di membrana ? 2
Canali ionici voltaggio-dipendenti: -proteine di membrana:la loro conformazione può modificarsi in conseguenza di variazioni del campo elettrico ai due lati della membrana: si può cosi’ passare da uno stato di impermeabilità ad uno di permeabilità selettiva per un dato ione. I movimenti transmembrana degli ioni attraverso specifici canali secondo gradienti di concentrazione ed elettrici, creando correnti ioniche a livello della membrana neuronale. L'alterazione in eccesso o in difetto di tali correnti contribuisce ai fenomeni di ipereccitabilità neuronale. Fondamentali sembrano le anomalie dei canali ionici voltaggio dipendenti di Na, Ca, K .

28
Meccanismi che regolano le proprietà di membrana ?3
Neuromediatori: vari sono i mediatori coinvolti Sembrano essere maggiormente coinvolti: l'acido g-aminobutirrico (GABA) ed il glutammato (GLU). -Il GABA è il neuromediatore inibitorio più diffuso nel SNC, essendo coivolto nei circuiti ricorrenti responsabili dei potenziali inibitori post-sinaptici (IPSP), che seguono i potenziali eccitatori. - L'inibizione GABAergica previene la risposta eccitatoria in risposta alla trasmissione sinaptica fisiologica. Due tipi di recettori per il GABA: GABA-A, che attiva un canale per il Cl, e GABA-B, che attiva un canale per il K.
 ed il glutammato (GLU). -Il GABA è il neuromediatore inibitorio più diffuso nel SNC, essendo coivolto nei circuiti ricorrenti responsabili dei potenziali inibitori post-sinaptici (IPSP), che seguono i potenziali eccitatori. - L inibizione GABAergica previene la risposta eccitatoria in risposta alla trasmissione sinaptica fisiologica. Due tipi di recettori per il GABA: GABA-A, che attiva un canale per il Cl, e GABA-B, che attiva un canale per il K.")
29
Meccanismi che regolano le proprietà di membrana ?3
Glutammato (GLU). -I recettori per il GLU sono estremamente rappresentati a livello dI'ippocampo e neocortex e sono accoppiati ad un canale per il Na: il legame del GLU con il suo recettore attiva una corrente Na inracellulare determinando un potenziale eccitatorio post-sinaptico (EPSP), che si caratterizza per una depolarizzazione graduale fino alla soglia del potenziale d'azione. Sono noti 3 tipi di recettori per il glutammato, definiti in base alla loro affinità per differenti agonisti del GLU
. -I recettori per il GLU sono estremamente rappresentati a livello dI ippocampo e neocortex e sono accoppiati ad un canale per il Na: il legame del GLU con il suo recettore attiva una corrente Na inracellulare determinando un potenziale eccitatorio post-sinaptico (EPSP), che si caratterizza per una depolarizzazione graduale fino alla soglia del potenziale d azione. Sono noti 3 tipi di recettori per il glutammato, definiti in base alla loro affinità per differenti agonisti del GLU.")
30
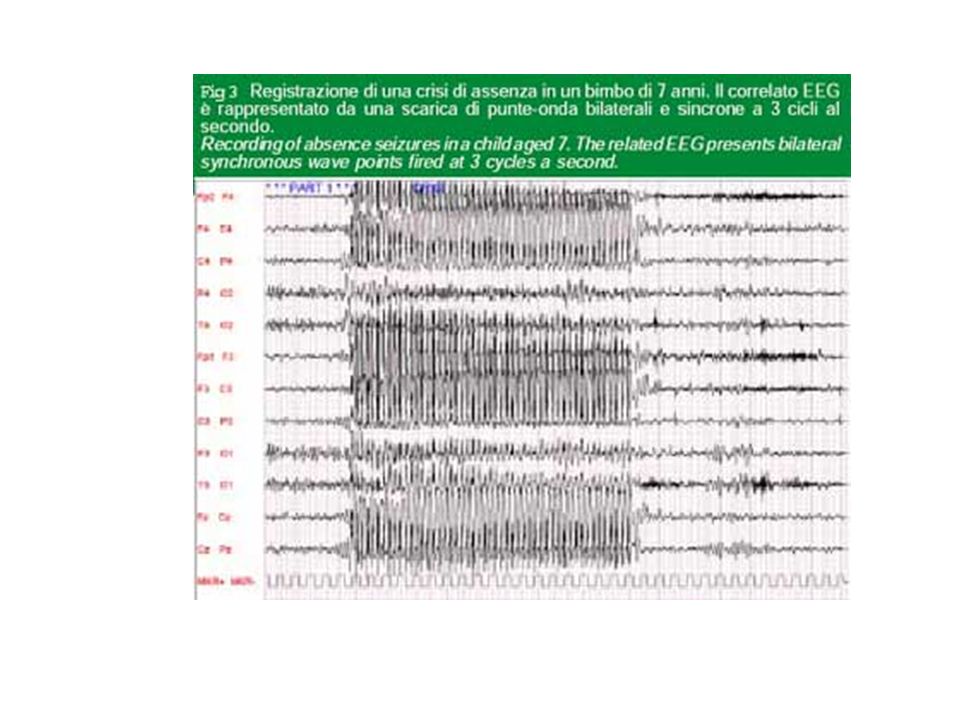
Nelle crisi generalizzate si ritiene che la scarica parossistica coinvolga fin dall'esordio e simultaneamente la corteccia di entrambi gli emisferi cerebrali. Nelle crisi a tipo assenza sono state riportate evidenze che implicano il coinvolgimento di un circuito riverberante con proiezioni reciproche cortico-talamiche e talamo-corticali. Tale circuito, che si attiva in maniera oscillante, comprende la corteccia ed i nuclei talamici di relais e reticolari.

33
Questo modello spiegherebbe l'efficacia del valproato e dell'etosuccimide che sopprimono le correnti mediate dai canali del Ca. Un modello di epilessia parziale, studiato anche su tessuto prelevato da pazienti, è quello delle epilessia con crisi temporali associate a sclerosi delle strutture temporali mesiali (STM). In questa condizione clinica, che si caratterizza dal punto di vista anatomico per una sclerosi ed una riduzione di volume delle strutture ippocampali, con conseguente perdita neuronale, i neuroni più suscettibili sono le cellule muscoidi (m) dell'ilo dentato
. In questa condizione clinica, che si caratterizza dal punto di vista anatomico per una sclerosi ed una riduzione di volume delle strutture ippocampali, con conseguente perdita neuronale, i neuroni più suscettibili sono le cellule muscoidi (m) dell ilo dentato.")
35
Genesi della una crisi epilettica focale?
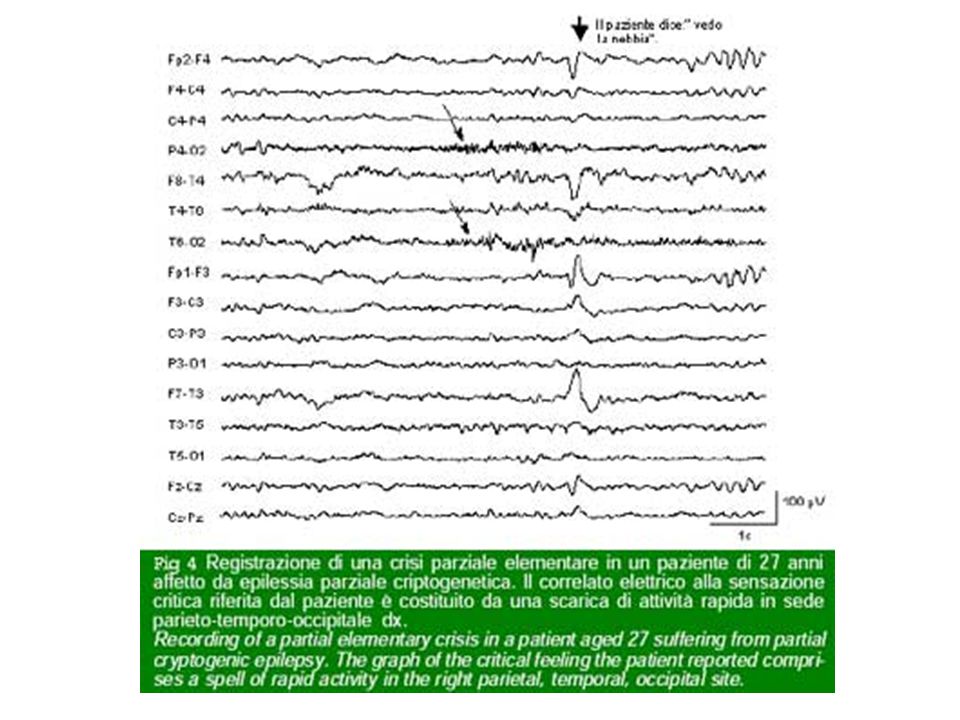
Le crisi focali originano da una regione limitata della corteccia cerebrale denominata zona epilettogena. Le manifestazioni cliniche ed EEG di una crisi dipendono dall'attivazione di circuiti epilettogeni, costituiti dalle diverse strutture cerebrali (anatomiche) sequenzialmente attivate dalla progressione della scarica epilettica originata dalla zona epilettogena. Le crisi temporali possono rappresentare un esempio di coinvolgimento di circuiti la cui complessità può variare a seconda del reclutamento di strutture diverse quali l'ippocampo, l'amigdala, la corteccia entorinale, la corteccia piriforme.
 sequenzialmente attivate dalla progressione della scarica epilettica originata dalla zona epilettogena. Le crisi temporali possono rappresentare un esempio di coinvolgimento di circuiti la cui complessità può variare a seconda del reclutamento di strutture diverse quali l ippocampo, l amigdala, la corteccia entorinale, la corteccia piriforme.")
36
La semeiologia di una crisi focale dipende dalle caratteristiche anatomo-funzionali del circuito epilettogeno attivata dalla scarica critica. Le manifestazioni cliniche dipendenti da una crisi focale possono variare da paziente a paziente, ma tendono ad essere stereotipe nello stesso paziente (se originano sempre dalla stessa zona epilettogena). Una crisi focale si caratterizza per la comparsa e successione temporale di una serie di manifestazioni che sono dipendenti dalla caratteristiche spazio-temporali di reclutamento delle strutture anatomiche attivate dalla scarica epilettica.
. Una crisi focale si caratterizza per la comparsa e successione temporale di una serie di manifestazioni che sono dipendenti dalla caratteristiche spazio-temporali di reclutamento delle strutture anatomiche attivate dalla scarica epilettica.")
38
Nella genesi delle crisi convulsive tonico-cloniche generalizzate vengono considerati meccanismi di tipo cortico-reticolare: la fase tonica dipenderebbe dall'attivazione di strutture sottocorticali, in particolare mesencefaliche;la fase clonica si realizzerebbe con la partecipazione della neocorteccia (probabilmente, anteriore).
.")
39
La terapia farmacologica.
Gli ultimi anni hanno visto l ’introduzione di diverse nuove molecole antiepilettiche, alcune delle quali presentano una migliore tollerabilità rispetto ai farmaci di vecchia generazione. Solo una quota modesta di pazienti resistenti ai farmaci di vecchia generazione ottengono il controllo completo delle crisi con i nuovi farmaci. Obiettivo primario della ricerca attuale è quello di identificare i meccanismi responsabili della farmacoresistenza e di sviluppare farmaci ancora più nuovi e caratterizzati da una maggiore efficacia e con minor

41
Summary of principal modes of action of AEDs

43
FARMACI E CITOCROMO P-450: livelli ridotti da farmaci induttori come CBZ, PB, DPH
Ormoni steroidei Acetominofene Ciclosporina, tacrolimus Doxiciclina Digossina Warfarin Teofillina Acido folico, vitamina D Neurolettici, antidepressivi triciclici Carbamazepina, Lamotrigina, Ac. Valproico

44
FARMACI che possono ridurre livelli plasmatici di Antiepilettici come Acido Valproico, Carbamazepina, Lamotrigina, Dintoina Interferenza con l’assorbimento Induzione enzimatica Antiacidi (anticipare la dose di 1-2 ore) Sucralfato Rifampicina Teofillina Carbamazepina, Fenobarbital,Dintoina
 Sucralfato. Rifampicina. Teofillina. Carbamazepina, Fenobarbital,Dintoina.")
45
FARMACI che possono aumentare i livelli plasmatici di Antiepilettici come Carbamazepina, Lamotrigina, Dintoina, Fenobarbital Inibizione enzimatica Cloramfenicolo,Eritromicina,Metronidazolo,Isoniazide Cimetidina (più degli altri H2 bloccanti) Diltiazem Verapamil Warfarin Fluoxetina Ac. Valproico
 Diltiazem. Verapamil. Warfarin. Fluoxetina. Ac. Valproico.")
46
Circa il 25% dei pazienti, nonostante la recente introduzione di numerosi fermaci aniticomiziali (AEDs),continua a presentare crisi. Kwan e Brodie (2000) e Arroyo et al (2002) definiscono l’epilessia farmacoresistente come una condizione in cui almeno due AEDs hanno fallito il controllo delle crisi, per una mancata efficacia, non per effetti collaterali. In tale situazione appare logico praticare politerapie che, più frequentemente comportano effetti negativi sul piano cognitivo e comportamentale o, in caso di focolai epilettogeni ben circoscritti, considerare l’approccio neurochirurgico. In alternativa appare opportuno valutare i diversi fattori che possono influenzare gli effetti farmacologici, con particolare attenzione sul ruolo di determinanti genetici sull’efficacia terapeutica.
 e Arroyo et al (2002) definiscono l’epilessia farmacoresistente come una condizione in cui almeno due AEDs hanno fallito il controllo delle crisi, per una mancata efficacia, non per effetti collaterali. In tale situazione appare logico praticare politerapie che, più frequentemente comportano effetti negativi sul piano cognitivo e comportamentale o, in caso di focolai epilettogeni ben circoscritti, considerare l’approccio neurochirurgico. In alternativa appare opportuno valutare i diversi fattori che possono influenzare gli effetti farmacologici, con particolare attenzione sul ruolo di determinanti genetici sull’efficacia terapeutica.")
47
Wiebe S et al N Engl J Med. 2001 Aug 2;345(5):311-8.
Randomized trials of surgery for epilepsy have not been conducted, because of the difficulties involved in designing and implementing feasible studies. Eighty patients with temporal-lobe epilepsy were randomly assigned to surgery (40 pts) or treatment with AEDs for one year (40 pts). At one year, the cumulative proportion of pts who were free of seizures impairing awareness was 58 % in the surgical group and 8 % in the medical group (P<0.001). The patients in the surgical group had fewer seizures impairing awareness and a significantly better quality of life than the pts in the medical group. Four pts (10 %) had adverse effects of surgery. CONCLUSIONS: In temporal-lobe epilepsy, surgery is superior to prolonged medical therapy.
 or treatment with AEDs for one year (40 pts). At one year, the cumulative proportion of pts who were free of seizures impairing awareness was 58 % in the surgical group and 8 % in the medical group (P<0.001). The patients in the surgical group had fewer seizures impairing awareness and a significantly better quality of life than the pts in the medical group. Four pts (10 %) had adverse effects of surgery. CONCLUSIONS: In temporal-lobe epilepsy, surgery is superior to prolonged medical therapy.")
48
Numerosi fattori possono influenzare gli effetti farmacologici,
la funzionalità renale ed , epatica, il concomitante uso di terapie, patologie concomitanti l’età Vi sono numerose evidenze che la differente risposta interindividuale nei confronti di uno stesso farmaco sia legata a varianti genetiche che agiscono a livello di: enzimi del metabolismo, trasportatori e recettori del farmaco (N Engl J Med, 2003).
.")
49
Assetto funzionale della citocromoossidasi P450 e dei suoi isoenzimi.
Le differenze genetiche di metabolizzazione sono importanti variabili interindividuali che fanno distinguere gli individui in lenti e rapidi metabolizzatori nella risposta individuale. Su tale concetto si basa il principio della farmacogenetica definita come lo studio del contributo genetico alla risposta farmacologica. Assetto funzionale della citocromoossidasi P450 e dei suoi isoenzimi.

50
MDR1 e MRP1 Tra i vari meccanismi di resistenza, l'iperespressione delle proteine della farmacoresistenza, come la glicoproteina-P1 (MDR1) e la proteina associata alla resistenza a più farmaci (MRP1), sono state mostrate essere correlate alla resistenza cellulare nei confronti dei farmaci antitumorali. Studi nell'epilessia hanno dimostrato che MDR1 e MRP1 possono anche essere iperespresse nel tessuto cerebrale dei pazienti con epilessia refrattaria, l'espressione è stata evidenziata nelle cellule gliali e nei neuroni, che normalmente non esprimono queste proteine.
 e la proteina associata alla resistenza a più farmaci (MRP1), sono state mostrate essere correlate alla resistenza cellulare nei confronti dei farmaci antitumorali. Studi nell epilessia hanno dimostrato che MDR1 e MRP1 possono anche essere iperespresse nel tessuto cerebrale dei pazienti con epilessia refrattaria, l espressione è stata evidenziata nelle cellule gliali e nei neuroni, che normalmente non esprimono queste proteine.")
51
Sviluppo della terapia chirurgica, grazie anche ai progressi della neuroradiologia.
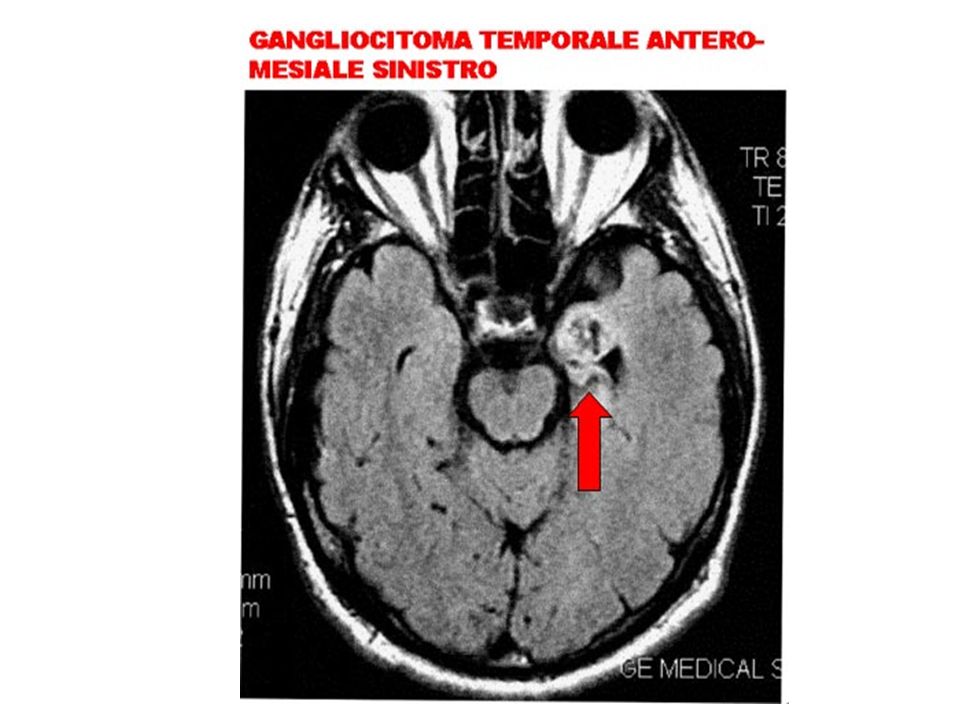
Si tratta della modalità terapeutica che si propone di asportare la zona di corteccia cerebrale coinvolta nella origine e propagazione della scarica. Si applica in casi di epilessia focale (ossia che ha origine da un’unica area ben definita del cervello) che non rispondono ai farmaci, selezionati in base a precise caratteristiche cliniche, neurofisiologiche e neuroradiologiche. Tale processo di selezione comporta la necessità di identificare, mediante tecniche innovative, l’ area da cui originano le crisi e la localizzazione delle aree nelle quali hanno sede le funzioni cognitive, sensoriali o motorie di ogni singolo malato
 che non rispondono ai farmaci, selezionati in base a precise caratteristiche cliniche, neurofisiologiche e neuroradiologiche. Tale processo di selezione comporta la necessità di identificare, mediante tecniche innovative, l’ area da cui originano le crisi e la localizzazione delle aree nelle quali hanno sede le funzioni cognitive, sensoriali o motorie di ogni singolo malato.")
52
La RMN cerebrale può essere considerata l'indagine di elezione per lo studio dell'epilessia. E' in grado di dimostrare alterazioni morfologiche in circa il 75% dei casi con TAC normale. Superiore risoluzione spaziale, Superiore definizione fra sostanza bianca e sostanza grigia, E’ in grado di rilevare lesioni isodense rispetto al tessuto cerebrale. E’in grado di rilevare piccole lesioni della base cranica, atrofia e sclerosi delle strutture ippocampali, displasie corticali ed alterazioni della migrazione neuronale, gliomi di piccole dimensioni e di basso grado, angiomi cavernosi, piccole lesioni ischemiche. L'esame è totalmente innocuo, pertanto non vi è alcuna controindicazione a ripeterlo più volte. Vi è la controindicazione per i pazienti portatori di clips metalliche oppure di pacemakers Vi sono difficoltà nell' effettuarlo in pazienti claustrofobici, o non cooperanti;

55
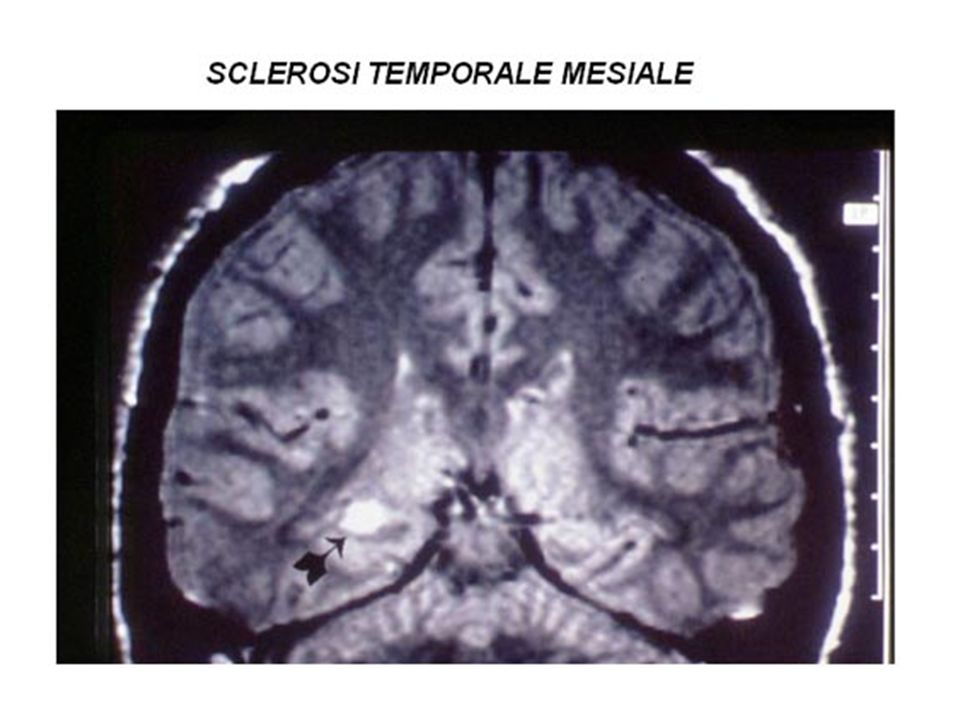
atrofia e sclerosi ippocampale
associate ad epilessia farmaco-resistente con crisi ad esordio delle strutture temporali antero-mesiali; si caratterizza per atrofia unilaterale di un ippocampo con dilatazione del corno temporale del ventricolo laterale contiguo, ipersegnale dell'ippocampo nelle sequenze coronali T2, riduzione di segnale nelle sequenze T1-pesate, e perdita della architettura normale nelle sequenze "inversion recovery".

57
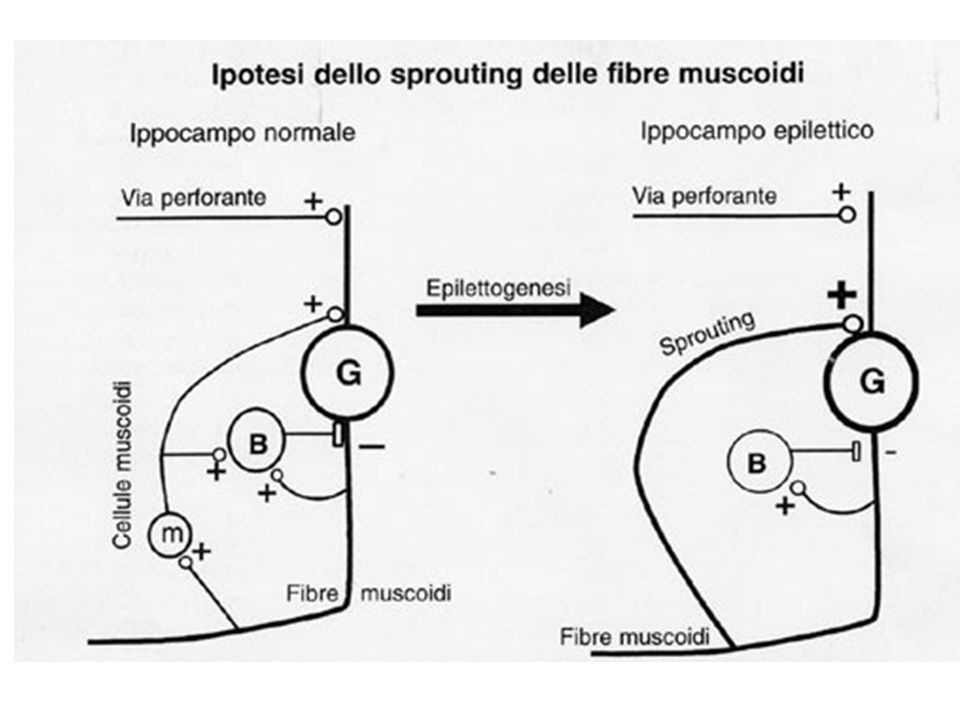
Le cellule m forniscono una innervazione eccitatoria cronica alle cellule a canestro GABA-ergiche, a funzione inibitoria. Con la morte delle cellule m manca una influenza inibitoria, mediata dalle cellule a canestro, e conseguente sviluppo di uno stato di ipereccitabilità. Inoltre, dalle cellule granulari dentate (G) originano sprouts, denominati fibre muscoidi (da non confondere con le cellule muscoidi) che si portano ai dendriti delle stesse cellule granulari: >>> circuito ricorrente autoeccitatorio, che favorisce lo sviluppo di proprietà epilettiche. Tale modello è sostenuto dall'evidenza che in ippocampi umani rimossi chirurgicamente è stato dimostrato istologicamente lo sprouting delle fibre muscoidi
 originano sprouts, denominati fibre muscoidi (da non confondere con le cellule muscoidi) che si portano ai dendriti delle stesse cellule granulari: >>> circuito ricorrente autoeccitatorio, che favorisce lo sviluppo di proprietà epilettiche. Tale modello è sostenuto dall evidenza che in ippocampi umani rimossi chirurgicamente è stato dimostrato istologicamente lo sprouting delle fibre muscoidi.")
58
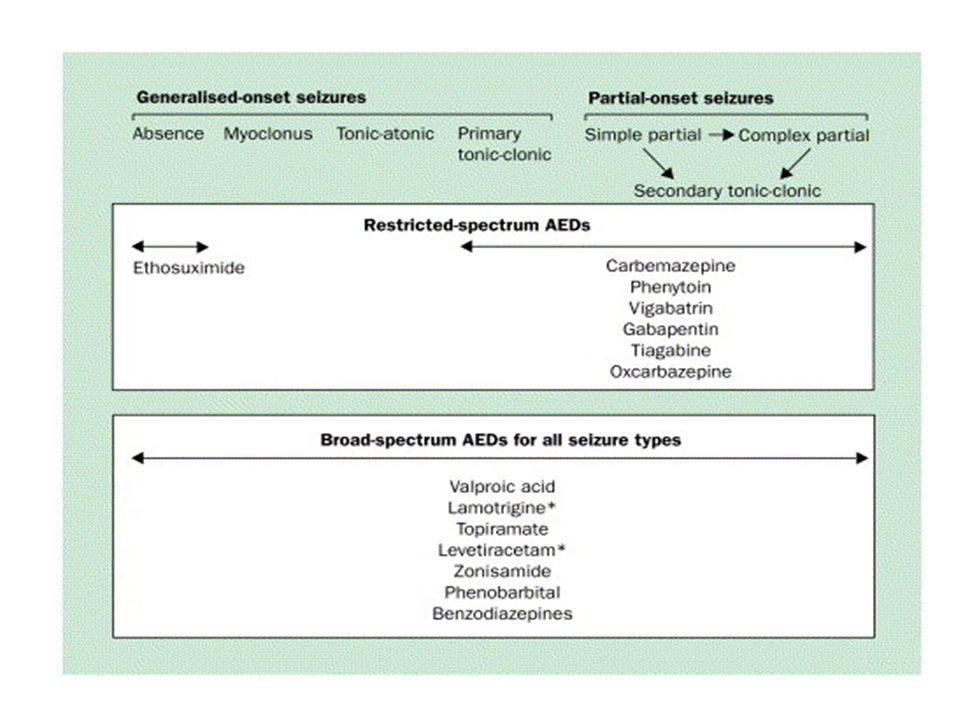
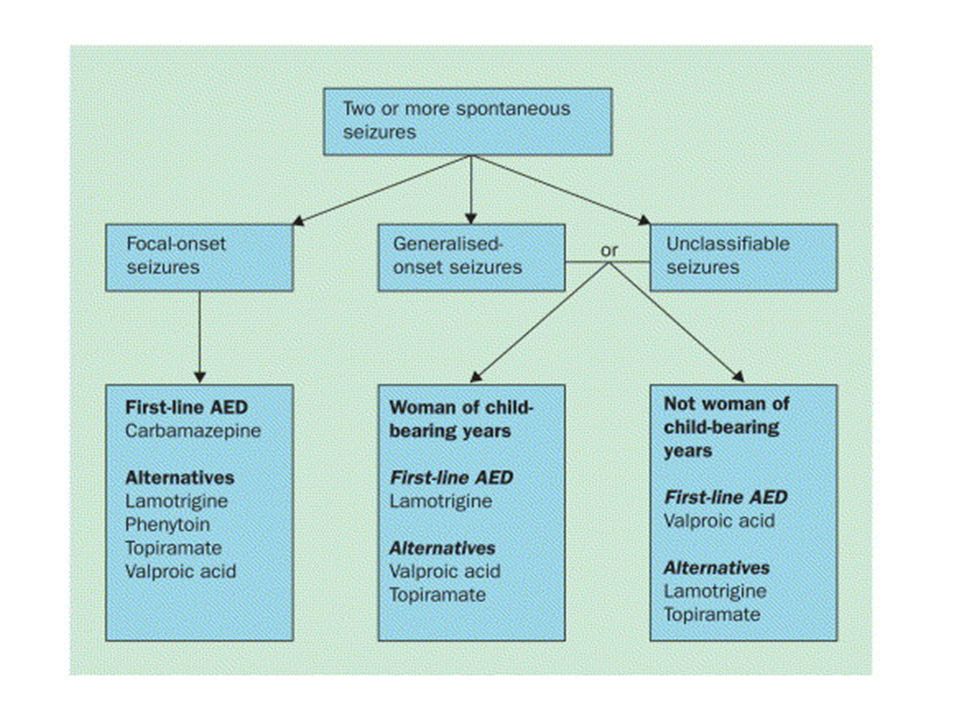
For refractory epilepsy, gabapentin, lamotrigine, topiramate, tiagabine, oxcarbazepine, levetiracetam, and zonisamide all have level A recommendations as adjunctive therapy (add-on to existing AED therapy) in adults with partial seizures. For children with partial seizures, there is evidence that gabapentin, lamotrigine, topiramate, and oxcarbazepine can be used (recommendation A). For refractory, primary generalized seizures only topiramate has a level A recommendation. For Lennox-Gastaut's syndrome, both lamotrigine and topiramate have level A recommendations as adjunctive therapy.
. For refractory, primary generalized seizures only topiramate has a level A recommendation. For Lennox-Gastaut s syndrome, both lamotrigine and topiramate have level A recommendations as adjunctive therapy.")
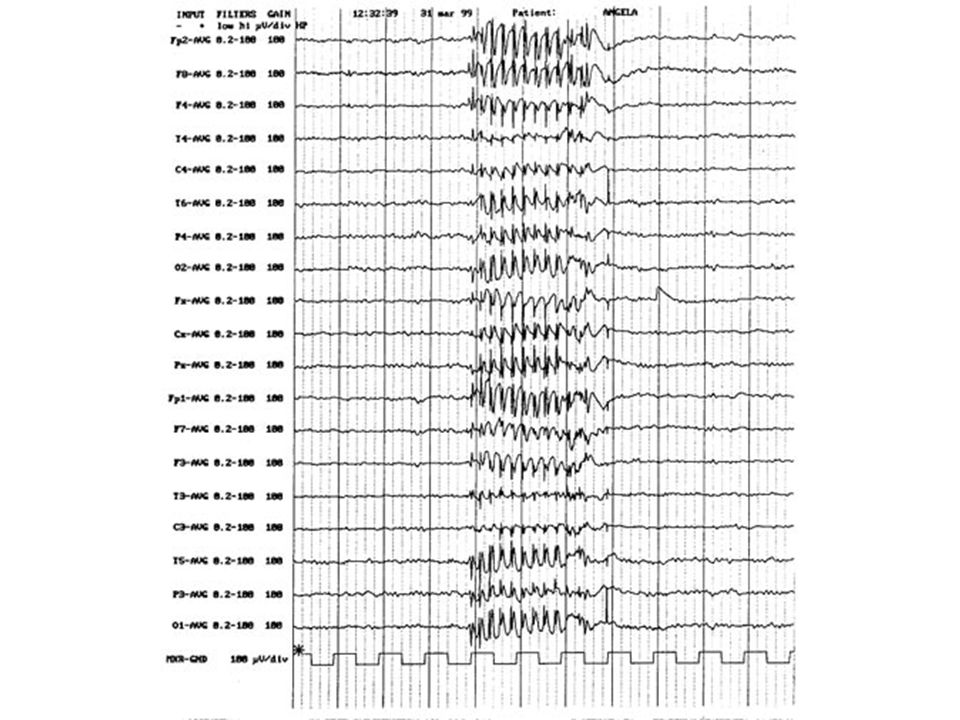
59
Esempio di crisi focale con secondaria generalizzazione

60
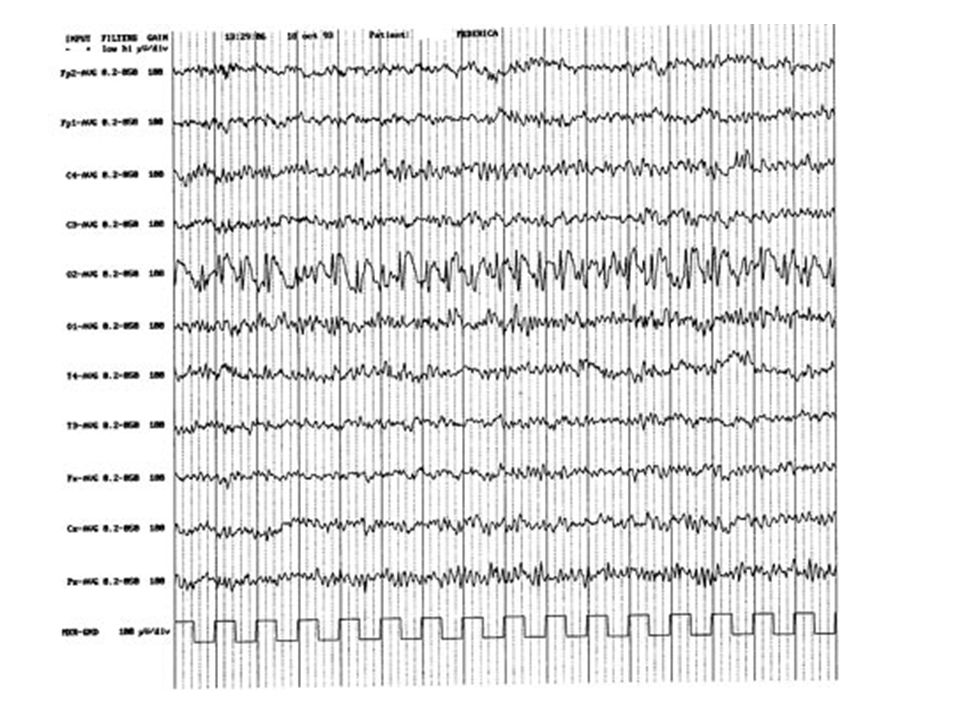
Circuiti cortico-talamici: Epilessia piccolo male
Le proiezioni cortico-talamiche sono reciprocamente eccitatorie con le proiezioni talamo-corticali, per proprietà oscillatorie intrinseche di questa regione. Neuroni GABA-ergici talamici reticolari tendono ad iperpolarizzare (mediante recettori GABA-B) i neuroni a proiezione talamo-corticale. I neuroni GABA-ergici B sono a loro volta inibiti da interneuroni GABA-ergici (attraverso recettori GABA-A). Entrambi i neuroni GABA-A e GABA-B infine ricevono innervazione dalla corteccia. Perché si verifichi la scarica talamo-corticale, secondaria all'attivazione cortico-talamica (e quindi si realizzi il meccanismo oscillatorio) occorre che i neuroni talamici presentino un'attivazione di canali T (talamici) del Ca, previa una iperpolarizzazione GABA-B mediata. La crisi di assenza pertanto può manifestarsi in conseguenza di una ipersensibilità dei canali T del Ca, dovuta ad una eccessiva iperattività GABA-B. Tale iperattività GABA-B può verificarsi solo se vi è una ridotta inibizione GABA-A a livello dei neuroni talamici reticolari
 i neuroni a proiezione talamo-corticale. I neuroni GABA-ergici B sono a loro volta inibiti da interneuroni GABA-ergici (attraverso recettori GABA-A). Entrambi i neuroni GABA-A e GABA-B infine ricevono. innervazione dalla corteccia. Perché si verifichi la scarica talamo-corticale, secondaria all attivazione cortico-talamica (e quindi si realizzi il meccanismo oscillatorio) occorre che i neuroni talamici presentino un attivazione di canali T (talamici) del Ca, previa una iperpolarizzazione GABA-B mediata. La crisi di assenza pertanto può manifestarsi in conseguenza di una ipersensibilità dei canali T del Ca, dovuta ad una eccessiva iperattività GABA-B. Tale iperattività GABA-B può verificarsi solo se vi è una ridotta inibizione GABA-A a livello dei neuroni talamici reticolari.")
Presentazioni simili




, sta ad indicare una modalità di reazione.>")


 (Ito) (IKr, IKs) (ICa-L) (INa).>")










>")




