Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
UNIVERSITA’ DEGLI STUDI DI FERRARA Dipartimento di Scienze Biomediche e Chirurgiche Specialistiche Sezione di Clinica Neurologica Aspetti clinici della malattia di Creutzfeldt-Jakob Prof Enrico Granieri. Anno Accademico 2015-2016

2
Primi casi descritti Creutzfeldt1920 ragazza di 22 anni Jakob 1921 Donna di 51 anni Donna di 34 anni Uomo di 42 anni

3
I fondatori della neurologia: Alfons Jakob 1884-1931

4
Clinica della malattia di Creutzfeldt-Jakob 1 Sintomi d’esordio: neuropsichici –Astenia –Ansietà –Turbe comportamento alimentare –Turbe del sonno –Perdita di peso Frequentemente l’esordio è subdolo e insidiosamente progressivo.

5
Clinica della malattia di Creutzfeldt-Jakob 2 Sintomi d’esordio neurologici: –Confusione mentale –Vertigine –Visione doppia –Tremori –Sensazione di instabilità –Incoordinazione dei gesti –Paresi –Formicolii, pruriti

6
Clinica della malattia di Creutzfeldt-Jakob decorso subacuto 1 Turbe comportamentali –Perdita memoria –Disorientamento spazio- temporale –Depressione dell’umore, ansietà, agitazione –Disordini afasici, aprassici, agnosici Turbe neurologiche primarie –Cerebellari Incoordinazione Disequilibrio Tremore intenzionale Disartria –Vie visive e visuomotorie Sdoppiamento immagini Cecità di parti di campo visivo Allucinazioni visive Mancato riconoscimento colori, distorsione delle forme

7
Clinica della malattia di Creutzfeldt-Jakob decorso subacuto 2 Aggravamento progressivo –Grave deterioramneto mentale –Grave incoordinazione motoria e perdita di equilibrio –Tremori, movimenti involontari patologici –Rigidità –Mioclonie brusche –(raramente) convulsioni Turbe vegetative Perdita di appetito e sete Episodi ricorrenti di ipertermia Ipersudorazione Sregolazione funzioni cardiovascolari
 convulsioni Turbe vegetative Perdita di appetito e sete Episodi ricorrenti di ipertermia Ipersudorazione Sregolazione funzioni cardiovascolari")
8
L’EEG nella malattia di Creutzfeldt Jakob

9
Electroencephalograms (EEGs) of two patients with sporadic CJD. Lower tracing shows the highly specific periodic sharp wave pattern; upper tracing shows a less specific but suggestive 'burst-suppression' pattern.

10
Magnetic resonance image (MRI) of a patient with sporadic Creutzfeldt-Jakob disease. The right basal ganglia (arrow) is abnormally bright. Usually, but not always, this abnormality is seen on both sides.
 is abnormally bright. Usually, but not always, this abnormality is seen on both sides..")
11
Clinica della malattia di Creutzfeldt-Jakob evoluzione Durata della malattia: evoluzione fino all’exitus in circa 1 anno –“Decorticazione” completa –Mutismo –Turbe della deglutizione –Cecità retinica e corticale Forme cliniche inabituali: Occipitali Talamiche Amiotrofiche Cerebellari Striato-nigriche

12
Encefalopatia spongiforme

13
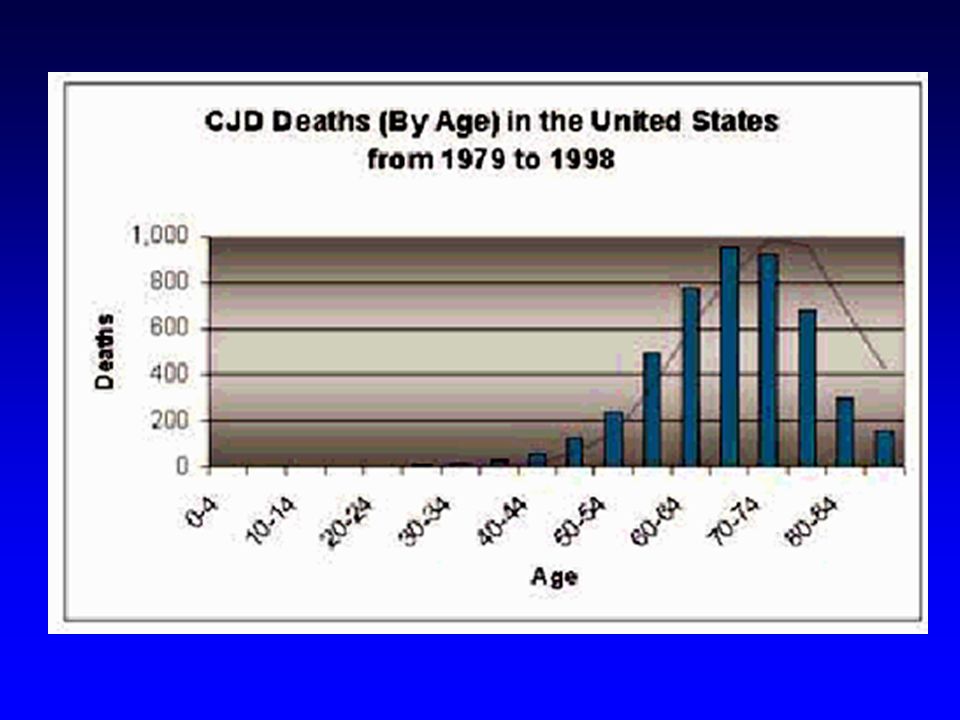
Tasso di incidenza per età in Europa 1993-1995

14
Tasso di mortalità in Europa 1993-1995, esclusi casi iatrogeni

16
Epidemia di BSE in U.K.

17
sCJD e nvCJD in Gran Bretagna Età all’esordio delle malattie (Will,2001)
")
18
Gajdusek, (Premio Nobel Medicina, 1976) 1966: omogenati di cervello di pazienti deceduti per CJD sporadica Macaca Mulatta 1968: diverse scimmie sviluppano i sintomi della malattia e presentano anomalie neuropatologiche simili alla malattia di Creutzfeldt-Jakob inoculazione Dopo 24-36 mesi
 1966: omogenati di cervello di pazienti deceduti per CJD sporadica Macaca Mulatta 1968: diverse scimmie sviluppano i sintomi della malattia e presentano anomalie neuropatologiche simili alla malattia di Creutzfeldt-Jakob inoculazione Dopo mesi")
19
Prione (PRusiner, 1982, Premio Nobel Medicina, 1997) Proteinaceus infectious particle Agenti proteici trasmissibili “non convenzionali” Insensibili ai trattamenti fisico-chimici inattivanti i virus Non inducono reazioni immunitarie
 Proteinaceus infectious particle Agenti proteici trasmissibili non convenzionali Insensibili ai trattamenti fisico-chimici inattivanti i virus Non inducono reazioni immunitarie")
20
PrP: Protease Resistant Protein

21
Criteri diagnostici per la sCJD European Criteria (Concerted action of EU,1994) 1. DEFINITE a.Neuropathological confirmation and/or b.PrP sc positive (Western blot) and/or c.SAF (Scrapie Associated Fibrils) 2. PROBABLE Progressive dementia At least 2 out of 4 clinical features listed: - myoclonus - visual or cerebellar - pyramidal/extrapyramidal - akinetic mutism Typical EEG or 14-3-3 in the CSF & duration less than 2 years 3. POSSIBLE Progressive dementia 2 out of clinical features listed above No EEG or atypical EEG Duration less than 2 years
 and/or c.SAF (Scrapie Associated Fibrils) 2. PROBABLE Progressive dementia At least 2 out of 4 clinical features listed: - myoclonus - visual or cerebellar - pyramidal/extrapyramidal - akinetic mutism Typical EEG or in the CSF & duration less than 2 years 3. POSSIBLE Progressive dementia 2 out of clinical features listed above No EEG or atypical EEG Duration less than 2 years.")
23
sCJD in Gran Bretagna Durata di malattia: in mesi maggio 1990-settembre 2001 (Will, 2001)
")
26
Malattie da prioni Caratteri comuni. –Lunga, lunghissima durata di incubazione asintomatica (per tutte le forme?) –Interessamento clinico degenerativo progressivo del Sistema Nervoso Centrale: Evoluzione rapida Comparsa costante di demenza associata a segni neurologici, soprattutto atassia Lesioni degenerative a carattere spongioso e proliferazione astrocitaria Accumulo di materiale prionico, anche a placche Assenza di reazioni infiammatorio Assenza di risposte immunitarie palesi Trasmissibilità, secondo modelli di infezioni all’animale d’esperimento sia per le forme sporadiche che per quelle genetiche ed acquisite
 –Interessamento clinico degenerativo progressivo del Sistema Nervoso Centrale: Evoluzione rapida Comparsa costante di demenza associata a segni neurologici, soprattutto atassia Lesioni degenerative a carattere spongioso e proliferazione astrocitaria Accumulo di materiale prionico, anche a placche Assenza di reazioni infiammatorio Assenza di risposte immunitarie palesi Trasmissibilità, secondo modelli di infezioni all’animale d’esperimento sia per le forme sporadiche che per quelle genetiche ed acquisite.")
27
Kuru Microscopically, one finds vacuolization in neuronal cytoplasm and dendrites, which gives the neuropil a spongy appearance

28
Encefalopatie spongiformi trasmissibili umane malattie da prioni Encefalopatia spongiforme trasmissibile sporadica –Malattia di Creutzfeldt-Jakob sporadica Encefalopatia spongiforme trasmissibile genetica –Malattia Creutzfeldt-Jakob familiare –Sindrome di Gerstmann Straüssler-Scheinker –Insonnia familiare fatale Encefalopatia spongiforme trasmissibile acquisita –Kuru –Malattia di Creutzfeldt-Jakob iatrogena –Malattia di Creutzfeldt-Jakob variante (inglese)
")
29
BSE

30
Variante inglese della malattia di Creutzfeldt-Jakob Tendenza all’esordio precoce: età 16-48 anni Debutto: neuro-psichico –Illusioni e allucinazioni uditive e visive –Depressione –Fenomeni dolorosi, pruriti, formicolii. Successivamente i fenomeni neurologici classici

31
clinica Disturbi psichiatrici (Depressione) Disturbi sensitivi (dolori e parestesie/Disestesie) quasi sempre agli arti Questi segni e sintomi precedono di diversi mesi: Disturbi d’equilibrio, Mioclonie e altre ipercinesie Perdita di memoria Poi… ulteriore evoluzione negativa: non si alimenta, non si muove, non parla,…
 Disturbi sensitivi (dolori e parestesie/Disestesie) quasi sempre agli arti Questi segni e sintomi precedono di diversi mesi: Disturbi d’equilibrio, Mioclonie e altre ipercinesie Perdita di memoria Poi… ulteriore evoluzione negativa: non si alimenta, non si muove, non parla,…")
32
Diagnosi RM: alterazioni nel talamo nella variante RM: alterazioni nel nucleo striato e nella corteccia nelle forme sporadiche Il prione si trova nelle tonsille

33
CJD variante

34
Final segmentation: grey matter (a), white matter (b), CSF without vessel class (c), T2 MRI (d), Proton Density MRI (e), CSF (f), other class including vessels (g). Zoom : Proton Density (h), CSF without vessel class (i), CSF (j), other class including vessels (k).
, CSF without vessel class (i), CSF (j), other class including vessels (k)..")
36
Epidemia di BSE in U.K.

37
Microscopically, spongy changes similar to those seen in kuru are found in the cortex. It is paramount when making the diagnosis of Creutzfeldt-Jakob disease that one is able to demonstrate that at least some of the vacuoles arise within neuronal cytoplasm. Other changes usually seen include red neurons and gliosis.

38
Placche di amiloide nella corteccia cerebrale (al centro), circondate da una zona di modificazione spongiforme Immunocitochimica per prion protein mostra una reazione fortemente positiva (marrone) in molte placche nella corteccia cerebrale
, circondate da una zona di modificazione spongiforme Immunocitochimica per prion protein mostra una reazione fortemente positiva (marrone) in molte placche nella corteccia cerebrale")
40
Nombre de cas de maladie de Creutzfeldt-Jakob Le Réseau national de surveillance des maladies de Creutzfeldt-Jakob et maladies apparentées met à jour la première semaine de chaque mois la statistique du nombre de décès par maladie de Creutzfeldt-Jakob survenus en France depuis 1992. Le nombre de cas probables de variante de la maladie de Creutzfeldt-Jakob non décédés figure aussi dans cette statistique. Les critères de diagnostic et la classification des différents types de maladie de Creutzfeldt-Jakob sont ceux du réseau européen EuroCJD.Le Réseau national de surveillance des maladies de Creutzfeldt-Jakob et maladies apparentéesLes critères de diagnostic et la classification Nombre de cas certains ou probables de MCJ en France par année de signalement pour les suspicions, par année de décès pour les MCJ décédées Mise à jour du 4 avril 2005

41
Polimormismo del gene PrP in Gran Bretagna Codone Popolazione sCJD nvCJD 129 normale M/M37% 82% 100% M/V 51% 10% 0% V/V 12% 8% 0%

42
Concetto di Barriera di Specie Difficoltà o impossibilità di trasmettere la malattia in specie animali (mammiferi) che differiscono dalla specie da cui deriva il materiale infetto (prion donor). Basi della barriera di specie: Differenze fisico-chimiche tra PrP sc del donatore e PrP c cellulare del ricevente (sequenza amino- acidica, conformazione, distribuzione delle cariche di superficie) Specificità di specie di altri fattori importanti nella conversione della PrP da forma normale (PrP c ) a forma patologica (PrP sc ).
 Specificità di specie di altri fattori importanti nella conversione della PrP da forma normale (PrP c ) a forma patologica (PrP sc )..")
43
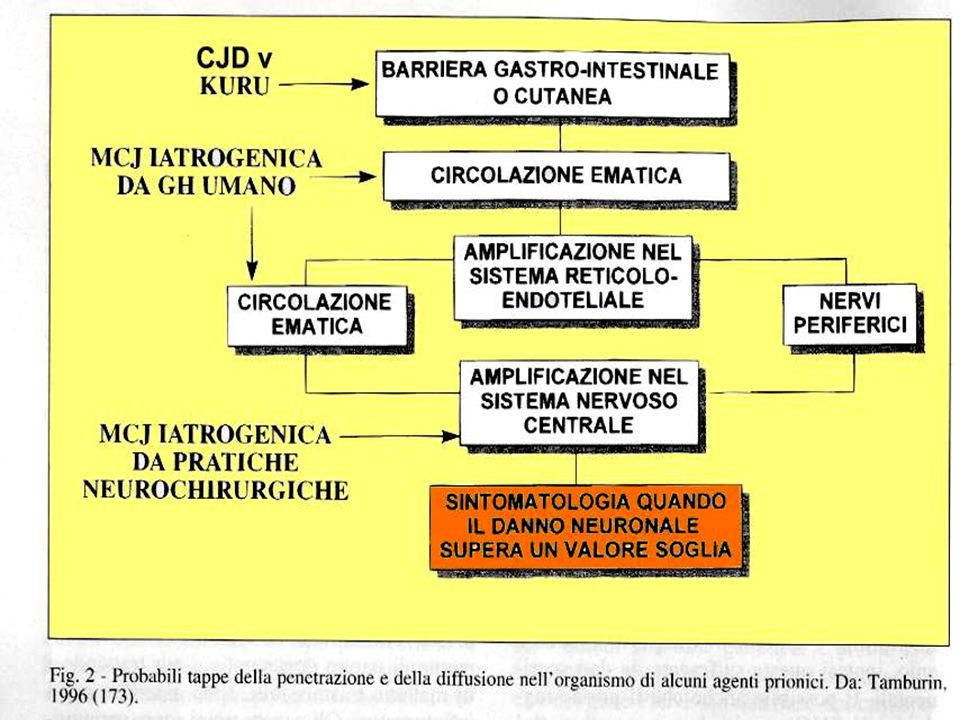
La trasmissione delle infezioni

45
CJD: sorveglianza epidemiologica europea Protocollo dello studio: La raccolta dei dati avviene tramite la somministrazione di un questionario, identico per i casi ed i controlli, costituito da domande chiuse. Esso viene somministrato al coniuge del paziente, o, in sua assenza, al parente più prossimo in grado di surrogarlo.

46
CJD: sorveglianza epidemiologica europea Il questionario si articola in sei sezioni dedicate alla raccolta delle informazioni anarnnestiche: 1)sezione demografica: dati demografici, geografici e socio-economici, con segnalazione di residenza rurale e di eventuali migrazioni; attività lavorative svolte. 2)sezione anamnesi familiare: storia familiare, con particolare riguardo alla patologia cerebrale involutiva ed alle attività lavorative dei genitori e del coniuge. 3)sezione abitudini di vita: viaggi e permanenze all' estero, in particolare in Gran Bretagna dopo il 1985; abitudini dietetiche, con particolare riferimento al consumo di carni e derivati, alla frequenza e al tipo di carni consumate; contatti con animali.
sezione anamnesi familiare: storia familiare, con particolare riguardo alla patologia cerebrale involutiva ed alle attività lavorative dei genitori e del coniuge. 3)sezione abitudini di vita: viaggi e permanenze all estero, in particolare in Gran Bretagna dopo il 1985; abitudini dietetiche, con particolare riferimento al consumo di carni e derivati, alla frequenza e al tipo di carni consumate; contatti con animali..")
47
CJD: sorveglianza epidemiologica europea 4)sezione anamnesi patologica: pregressi interventi chirugici, in particolare neurochirurgici, con eventuale innesto di dura madre; trapianti, in particolare di cornea; interventi oftalmologici e tonometria oculare; traumi cranici; malattie infettive pregresse; trasfusioni; impianto di elettrodi per EEG stereotassica. 5)sezione anamnesi farmacologica: indagini su pregresse vaccinazioni; terapie iniettive, in particolare con principi attivi di estrazione o di derivazione animale; terapie ormonali estrattive. 6)un 'ultima sezione fornisce informazioni su altre esposizioni a fattori di rischio biologicamente plausibili quali agopuntura, tatuaggi, foro all’orecchio, elettromiografia
sezione anamnesi farmacologica: indagini su pregresse vaccinazioni; terapie iniettive, in particolare con principi attivi di estrazione o di derivazione animale; terapie ormonali estrattive. 6)un ultima sezione fornisce informazioni su altre esposizioni a fattori di rischio biologicamente plausibili quali agopuntura, tatuaggi, foro all’orecchio, elettromiografia.")
50
terapie Farmaci in sperimentazione per prolungare il decorso della malattia e la qualità di vita del paziente. QUINACRINA: vecchio antimalarico PENTOSAN POLISOLFATO: interagisce con la coagulazione del sangue DOXICLINA: antibiotico che si lega alla proteina prionica e la rende aggredibile

51
sporadica Malattie da prioni Differenze cliniche talora significative Variabilità neuropatologica Epidemiologia diversa. –Forma sporadica –Forma genetica –Forma iatrogenica –Forme varianti alimentari prioni genetica iatrogenica varianti alimentari

52
Particolari precauzioni oggi adottate In laboratorio Nelle sale di autopsia In ambienti chirurgici Negli ospedali Nei casi di ospedalizzazione di pazienti con malattia di Creutzfeldt-Jakob

53
L’individuazione delle Encefalopatie Trasmissibili, o malattie da Prioni, ha il merito, indipendentemente dall’interesse scientifico e medico (che resta prioritario), di richiamare l’at- tenzione sul danno potenziale che potrebbe emergere nella disinvolta manipolazione di prodotti di origine animale e umana e sulla urgente necessità di programmare l’utilizzazione in maniera ragionata, prudente e parsimoniosa
, di richiamare l’at- tenzione sul danno potenziale che potrebbe emergere nella disinvolta manipolazione di prodotti di origine animale e umana e sulla urgente necessità di programmare l’utilizzazione in maniera ragionata, prudente e parsimoniosa")
54
“TERAPIE ANTIPRIONICHE” Antracicline, Rosso Congo Solfato destrano Pentosan Polisulfato Polianioni B-sheet breaker peptidi Efficaci solo se somministrati prima dell’inizio della malattia Alti livelli di tossicità Bassi livelli di biodisponibilità

55
Terapie antiprioniche Sono in corso clinical trials Quinacrina Clorpromazina Non c’è ancora evidenza di efficacia In UK è stata imposta l’inoculazione intra- ventricolare di di pentosano polisolfato a due pazienti..

56
Sindrome di Gerstmnn-Straussler- Scheinker Autosomica dominante con penetranza completa Reports nell’Emisfero Nord Incidenza: 1-10 per 100,000,000/anno (è sottostimata?) Esordio età adulta: 40-45 anni Durata di malattia: 4-11 anni Criteri classificativi incerti per eterogeneità nell’espressione clinica Peculiarità genetiche (mutazioni ai codoni 102,117,…) Peculiarità neuropatologiche (Placche di amiloide multicentriche cerebrali e cerebellari,..) Spontanea in topi transgenici Infezione trasmessa da inoculazione di GSSD-brain
 Esordio età adulta: anni Durata di malattia: 4-11 anni Criteri classificativi incerti per eterogeneità nell’espressione clinica Peculiarità genetiche (mutazioni ai codoni 102,117,…) Peculiarità neuropatologiche (Placche di amiloide multicentriche cerebrali e cerebellari,..) Spontanea in topi transgenici Infezione trasmessa da inoculazione di GSSD-brain")
57
INSONNIA FAMILIARE FATALE Malattia autosomica dominante Incidenza sconosciuta Esordio: 35-61 anni di età Eterogeneità clinica: insonnia intrattabile, segni motori, Durata di malattia: 12 mesi (7-25 mesi) Peculiarità neuropatologiche: atrofia dei nuclei talamici, Peculiarità genetiche (mutazione del codone 178, asparagina sostituisce acido aspartico..)
 Peculiarità neuropatologiche: atrofia dei nuclei talamici, Peculiarità genetiche (mutazione del codone 178, asparagina sostituisce acido aspartico..)")
Presentazioni simili

















>")


 Casi clinici>")





