Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
YT

2
Media Type Chemically Defined Rich Media Composition Composition
Buffer (phosphate based) Nitrogen source (ammonium chloride or ammonium sulfate) Carbon Source (glucose or glycerol) Trace Metals (Ca, Co, Cu, Fe, Mn, Mo, Se and Zn) Vitamins (Biotin, thiamine, folic acid, riboflavin, niacinamide, PABA, pantothenic acid and pyridoxine) Composition Tryptone Yeast extract Sodium Chloride Examples Luria Broth (LB) 2xYT TB (phosphate buffered)
 Nitrogen source (ammonium chloride or ammonium sulfate) Carbon Source (glucose or glycerol) Trace Metals (Ca, Co, Cu, Fe, Mn, Mo, Se and Zn) Vitamins (Biotin, thiamine, folic acid, riboflavin, niacinamide, PABA, pantothenic acid and pyridoxine) Composition. Tryptone. Yeast extract. Sodium Chloride. Examples. Luria Broth (LB) 2xYT. TB (phosphate buffered)")
3
M9 minimal medium M9 medium is a minimal growth medium used for bacterial cultures. It has the advantage of being cheap 1x M9 salts 2mM MgSO4 0.1mM CaCl2 0.4% carbon source (e.g. glycerol, glucose, etc.) 1g nitrogen source (15N) In sterile H2O 5X M9 salts contains: 64 g Na2HPO4.7H2O 15 g KH2PO4 2.5 g NaCl 5 g NH4Cl dissolved in deionized water to a final volume of 1 L Isotopically labeled proteins can be prepared straightforwardly in E. coli by growing cells in minimal media M9 supplemented with appropriate nutrients (15NH4Cl, 13C-glucose) For large proteins deuterium labeling provides simplified spectra for the remaining 1H nuclei and has useful effects on relaxation properties of attached or adjacent atoms (1H, 15N,13C).
 1g nitrogen source (15N) In sterile H2O. 5X M9 salts contains: 64 g Na2HPO4.7H2O. 15 g KH2PO g NaCl. 5 g NH4Cl. dissolved in deionized water to a final volume of 1 L. Isotopically labeled proteins can be prepared straightforwardly in E. coli by growing cells in minimal media M9 supplemented with appropriate nutrients (15NH4Cl, 13C-glucose) For large proteins deuterium labeling provides simplified spectra. for the remaining 1H nuclei and has useful effects on relaxation. properties of attached or adjacent atoms (1H, 15N,13C).")
4
Rich vs. Minimal Media pQE expression of At3g17210
pET expression of At3g17210

5
Parametri delle colture

7
Temperatura Da 16°C a 37°C Surf1 pTH24 C41 25° Surf1 pTH24 C41 18°
M IB IB Prot FT1 FT2 W1 W2 W3 W4 W5 E1 E2 E3 M IB Prot FT1 FT2 W1 W2 W3 W4 W5 E1 E2 E3

9
Differenti concentrazioni Induzioni a tempi diversi
IPTG Differenti concentrazioni 1mM - 0.1mM Induzioni a tempi diversi

10
t Unità formanti colonie latenza esponenziale plateau morte FASE DI LATENZA: intervallo di tempo in cui i microrganismi si adattano all’ambiente (non si moltiplicano) FASE ESPONENZIALE: periodo di tempo in cui i microrganismi si moltiplicano in modo progressivo e costante PLATEAU: si stabilisce un equilibrio tra il numero delle cellule che si moltiplicano e il numero di cellule che muoiono FASE DELLA MORTE: cessa la moltiplicazione dei microrganismi che invecchiano e muoiono
 FASE ESPONENZIALE: periodo di tempo in cui i microrganismi si moltiplicano in modo progressivo e costante. PLATEAU: si stabilisce un equilibrio tra il numero delle cellule che si moltiplicano e il numero di cellule che muoiono. FASE DELLA MORTE: cessa la moltiplicazione dei microrganismi che invecchiano e muoiono.")
11
Trasformazione del DNA per espressione proteica
Trasformazione in BL21 A l di cellule competenti di E. coli (BL21) si aggiungono X l (corrispondenti a 100 ng) di uno dei campioni di DNA plasmidico che hanno dato risultato positivo allo screening. Controllo: in parallelo compiere la stessa reazione aggiungendo X l di acqua sterile invece del plasmide. 30’, ghiaccio 90’’, 42°C 2’, ghiaccio aggiungere l 2xYT, 1h shaker a 37°C, 150 rpm. Strisciare su piastre contenenti 2xYT + amp (200 g/ml), l di ognuna delle trasformazioni. O/N, 37°C
 si aggiungono. X l (corrispondenti a 100 ng) di uno dei campioni di DNA plasmidico. che hanno dato risultato positivo allo screening. Controllo: in parallelo compiere la stessa reazione aggiungendo X l di. acqua sterile invece del plasmide. 30’, ghiaccio. 90’’, 42°C. 2’, ghiaccio. aggiungere l 2xYT, 1h shaker a 37°C, 150 rpm. Strisciare su piastre contenenti 2xYT + amp (200 g/ml), l di. ognuna delle trasformazioni. O/N, 37°C.")
12
Coltura massiva Inoculo
Si prelevano singole colonie che vengono trasferite in alcuni ml di terreno liquido. Si lascia incubare in shaker alla temp. opportuna per il ceppo batterico, per circa ore (fase stazionaria) Coltura massiva L’inoculo viene trasferito in alcuni litri di terreno liquido, 1% in volume . Si lascia incubare in shaker alla temp. opportuna. All’inizio della fase esponenziale l’espressione della proteina viene indotta per aggiunta di un induttore che determina l’avvio della trascrizione proteica 4 8 12 16 fase esponenziale Abs600 +IPTG raccolta cellule tempo di incubazione IPTG
 Coltura massiva. L’inoculo viene trasferito in alcuni litri di terreno liquido, 1% in volume . Si lascia incubare in shaker alla temp. opportuna. All’inizio della fase esponenziale l’espressione della proteina viene indotta per aggiunta. di un induttore che determina l’avvio della trascrizione proteica fase esponenziale. Abs600. +IPTG. raccolta cellule. tempo di incubazione. IPTG.")
13
Procedura per l’espressione di rame proteina
Preparazione inoculo da piastra Inoculare 2 tubi contenenti ciascuno 6 ml di 2xYT + amp (200 g/ml) con 2-3 colonie prelevata da piastra. Mettere ad incubare in shaker O/N a 37°C. Preparazione coltura massiva Inoculare 2 beute da 2l contenenti ciascuna 0.5 l 2xYT + amp (200 g/ml) con 5 ml di inoculo già preparato. Mettere ad incubare in shaker a 37°C. Misurare ad intervalli regolari la densità ottica a 600 nm. Quando la densità ottica raggiunge il valore di 0.6, aggiungere a ciascuna beuta 1.0 ml di soluzione di IPTG 0.5 M (concentrazione finale 1 mM) e 125 l di soluzione di CuSO4 1 M Continuare l’incubazione in shaker alla temperatura di 25°C per 16 h. Aggiungere 375 l di soluzione di CuSO4 1 M 45’ prima di raccogliere le cellule Isolamento della proteina Rottura delle cellule Separazione dell’estratto dalle cellule
 con 2-3 colonie. prelevata da piastra. Mettere ad incubare in shaker O/N a 37°C. Preparazione coltura massiva. Inoculare 2 beute da 2l contenenti ciascuna 0.5 l 2xYT + amp (200 g/ml) con 5 ml di inoculo. già preparato. Mettere ad incubare in shaker a 37°C. Misurare ad intervalli regolari la densità ottica a 600 nm. Quando la densità ottica raggiunge il valore di 0.6, aggiungere a ciascuna beuta 1.0 ml di. soluzione di IPTG 0.5 M (concentrazione finale 1 mM) e 125 l di soluzione di CuSO4 1 M. Continuare l’incubazione in shaker alla temperatura di 25°C per 16 h. Aggiungere 375 l di soluzione di CuSO4 1 M 45’ prima di raccogliere le cellule. Isolamento della proteina. Rottura delle cellule. Separazione dell’estratto dalle cellule.")
18
Ottimizzazione per lo scale-up

19
Minifors fermenters Interactive control of culture parameters: Temp, pH, pO2 Parameters can be automatically changed during the culture from a computer Possibility of feed/continuous cultures

20
GESTIONE DEI PARAMETRI DI PROCESSO
Temperatura riscaldamento/raffreddamento Agitazione impostazione/variazione di un “set point” Flusso aria pH aggiunte di acido/base e/o feed pO2 agitazione/ flusso aria/ arricchimento in O2 e/o feed Schiuma aggiunte di antischiuma Alimentazione impostazione/variazione di un ciclo di attività della pompa del feed

22
Nei processi di estrazione e purificazione delle proteine, a differenza di quanto avviene per il DNA/cellule, non occorre prestare attenzione alla sterilità ma bisogna evitare la denaturazione dei campioni (elevate temperature, detergenti, valori estremi di pH possono denaturare facilmente le proteine) Inoltre è importate procedere all’estrazione delle proteine il più velocemente possibile, per evitare processi degradativi. Per conservare i campioni da cui estrarre le proteine vanno congelati rapidamente in azoto liquido (-140°C) e poi conservati in frigo a -80°C
 e poi conservati in frigo a -80°C.")
23
ESTRAZIONE______________________________________________
LISI DELLE CELLULE Localizzazione della proteina Tipo di cellule Quantità di cellule BUFFER DI LISI Parametri chimico-fisici della proteina (pI, idrofobicità, …) Presenza di Cys ridotte/ossidate Metalli Regioni transmembrana Attività proteolitica Rimozione di acidi nucleici CHIARIFICAZIONE Centrifugazione Ultrafiltrazione
 Presenza di Cys ridotte/ossidate. Metalli. Regioni transmembrana. Attività proteolitica. Rimozione di acidi nucleici. CHIARIFICAZIONE. Centrifugazione. Ultrafiltrazione.")
24
Proteine extracellulari secrete nel terreno di cultura o nel siero possono essere rimosse dal materiale insolubile per semplice centrifugazione Proteine intracellulari possono essere recuperate dopo rottura delle cellule La procedura adottate per ottenere un estratto grezzo (estratto contenente tutte le proteine cellulari solubili nel tampone di estrazione utilizzato) dipende dalla localizzazione cellulare della proteina di interesse.
 dipende dalla localizzazione cellulare della proteina di interesse.")
26
I metodi utilizzati per la rottura delle cellule dipendono in gran parte dalla fragilità delle cellule Cellule animali -Cellule fragili (eritrociti) -Cellule che contengono materiale fibroso (c. muscolari) Cellule vegetali Più difficili da rompere perché contengono cellulosa Microorganismi Batteri che si lisano facilmente Batteri con pareti più resistenti
 -Cellule che contengono materiale fibroso. (c. muscolari) Cellule vegetali. Più difficili da rompere perché contengono cellulosa. Microorganismi. Batteri che si lisano facilmente. Batteri con pareti più resistenti.")
27
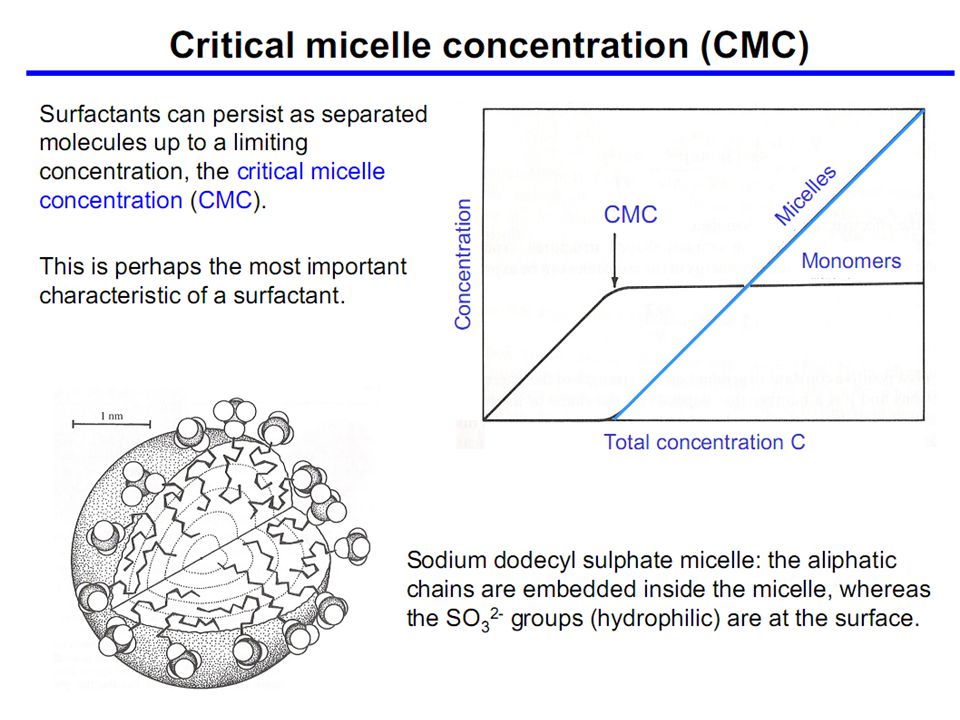
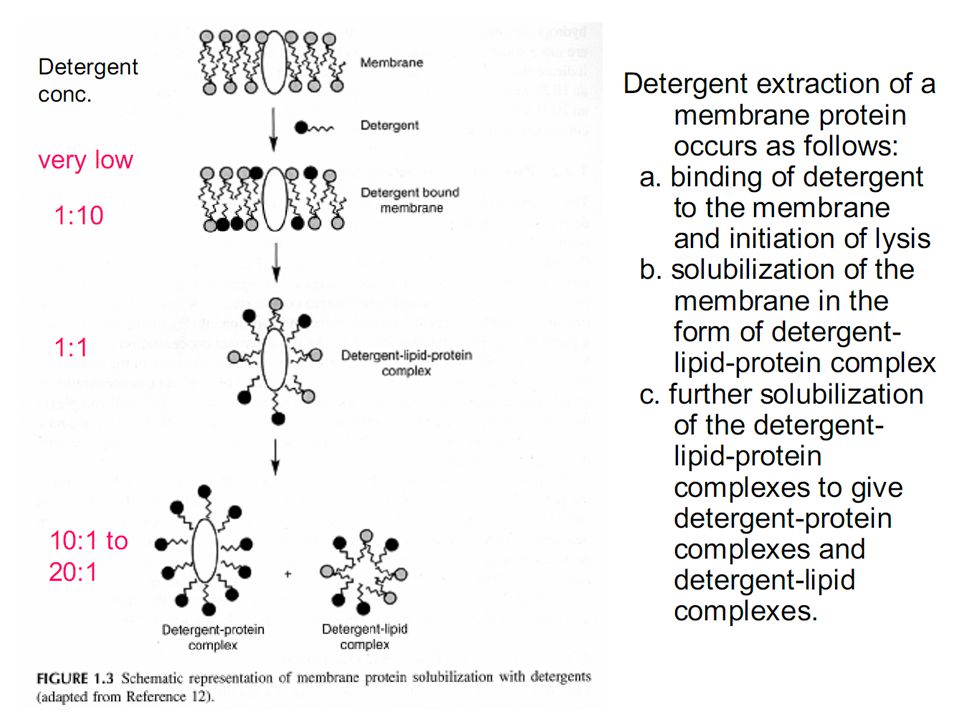
Utilizzo di detergenti per dissolvere
la membrana cellulare

28
Proteine di secrezione
Le proteine contengono segnali che ne determinano la destinazione. Una proteina può restare nel citosol, può essere portata sulle membrane plasmatica o interna, nello spazio intermembrana o nel mezzo intracellulare. Sequenze segnale aminoterminali (16-26 AA) dirigono le proteine batteriche attraverso le membrane plasmatiche od esterne del batterio. Vengono riconosciute e tagliate da opportune peptidasi Proteine secrete nel periplasma possono essere recuperate per rottura della sola membrana esterna (shock osmotico)
 dirigono le proteine batteriche attraverso le. membrane plasmatiche od esterne del batterio. Vengono riconosciute e tagliate da opportune peptidasi. Proteine secrete nel periplasma possono essere recuperate per rottura. della sola membrana esterna (shock osmotico)")
29
Shock osmotico Per 1 litro di coltura
Centrifugare la cultura a 8000 rpm per 10’, 4°C. Disperdere il pellet in 50 ml Tris-HCl 10 mM a pH 7.5. Aggiungere 50 ml di soluzione di saccarosio al 40% in 10 mM Tris-HCl pH ml di soluzione EDTA 0.5 mM pH 8. Le soluzioni devono essere ice-cold. Tenere in agitazione in ghiaccio in camera fredda per 20’. Centrifugare rpm per 15’ a 4°C. Rimuovere accuratamente il surnatante. Disperdere il pellet in 30 ml H2O fredda tenere in bagno di ghiaccio per 20’. Centrifugare per rpm per 15‘ a 4°C. Raccogliere accuratamente il surnatante che contiene la proteina. Disperdere nuovamente il pellet in 20 ml H2O fredda e tenere ancora per 20’ in bagno di ghiaccio sotto agitazione. Centrifugare a rpm per 15’ a 4°C e raccogliere il surnatante. Riunire i due surnatanti.

31
LISI DELLE CELLULE

32
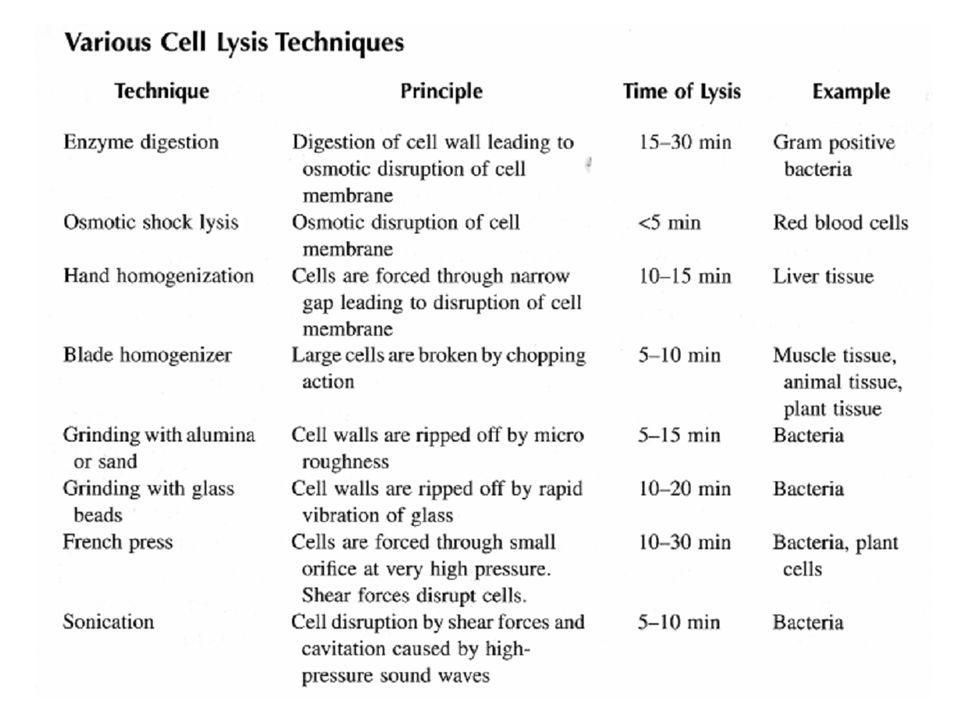
Tecniche di rottura delle cellule

33
Uso del lisozima nella rottura della parete cellulare
Il lisozima lisa alcuni tipi di batteri rompendo la componente polisaccaridica della parete, formata da 2 tipi di zuccheri, N-acetilglucosamina (NAM) e da acido N-acetilmuranico (NAG). La parete perde rigidita’ e il batterio scoppia a causa dell’elevata pressione osmotica intracellulare Il lisozima idrolizza il legame glicosidico tra il C-1 del NAM e il C-4 del NAG
 e da. acido N-acetilmuranico (NAG). La parete perde rigidita’ e il batterio scoppia a causa dell’elevata pressione osmotica. intracellulare. Il lisozima idrolizza il legame glicosidico tra il C-1 del NAM e il C-4 del NAG.")
34
Alcuni sistemi per rompere le cellule

35
Vari modelli di omogeneizzatore.

40
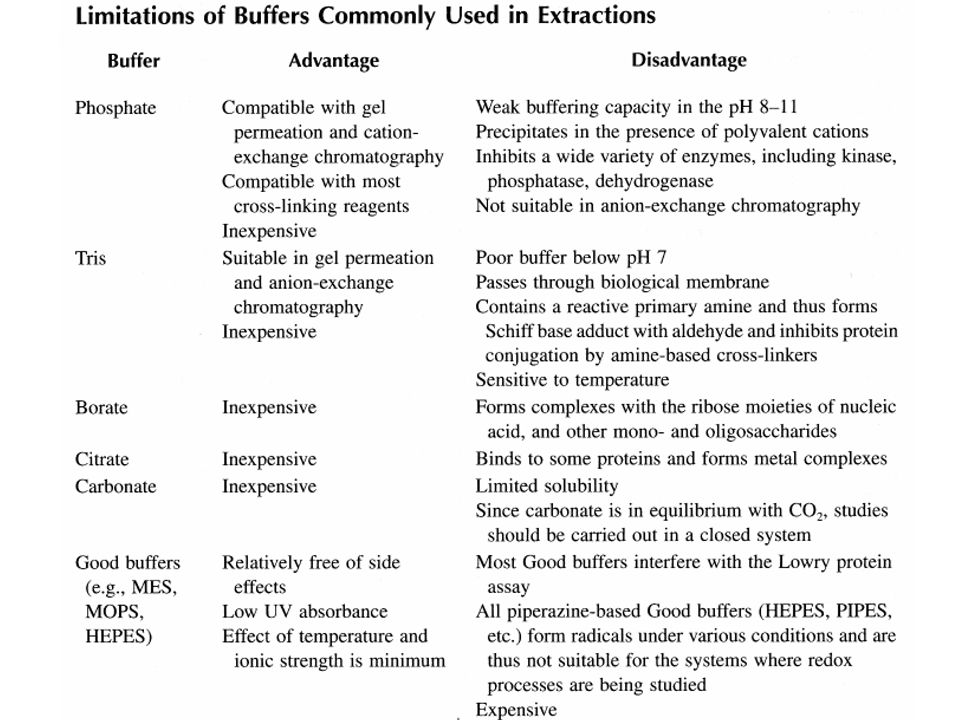
SCELTA DEL BUFFER DI LISI
I buffer più utilizzati sono: Fosfato – Tris – HEPES (pH: 5.8-9) Concentrazioni del buffer: mM Tris: il pH dipende dalla temperatura pKa: a 25°C 8.85 a 0°C
 Concentrazioni del buffer: mM. Tris: il pH dipende dalla temperatura pKa: 8.06 a 25°C 8.85 a 0°C.")
41
SCELTA DEL BUFFER DI LISI
pH range Citric acid - NaOH Sodium citrate - citric acid Sodium acetate - acetic acid Cacodylic acid sodium salt - HCl MES - NaOH Sodium dihydrogen phosphate - disodium hydrogen phosphate Imidazole - HCl MOPS - KOH Triethanolamine hydrochloride - NaOH Tris - HCl HEPES - NaOH Tricine - NaOH Sodium tetraborate - boric acid Bicine - NaOH Glycine - NaOH

42
TAMPONI COMMERCIALI

44
SCELTA DEL BUFFER DI LISI
ADDITIVI: Aumentano la stabilità della proteina Aumentano la solubilità della proteina Class of additive example concentration purpose Salts NaCl, KCl, (NH4)2SO4 mM maintain ionic strength of medium Detergents Deoxycholate, Triton X-100 0.1-1% solubilization of poorly soluble proteins Glycerol 5-10% stabilization Glucose or sucrose 25 mM Stabilize lysosymal membranes, reduce protease release Metal chelators EDTA, EGTA 1 mM reduce oxidation damage, chelate metal ions Reducing agents DTT, DTE 2-Mercaptoethanol 1-10 mM 0.05% reduce oxidation damage Ligands, metal ions Mg2+, ATP, GTP 1-10 mM Protease Inhibitors PMSF, Aprotinin,… Reduced protease activity DNase DNAse I U/ml Reduced solution viscosity
2SO mM. maintain ionic strength of medium. Detergents. Deoxycholate, Triton X % solubilization of poorly soluble proteins. Glycerol. 5-10% stabilization. Glucose or sucrose. 25 mM. Stabilize lysosymal membranes, reduce protease release. Metal chelators. EDTA, EGTA. 1 mM. reduce oxidation damage, chelate metal ions. Reducing agents. DTT, DTE 2-Mercaptoethanol mM 0.05% reduce oxidation damage. Ligands, metal ions. Mg2+, ATP, GTP mM. Protease Inhibitors. PMSF, Aprotinin,… Reduced protease activity. DNase. DNAse I U/ml. Reduced solution viscosity.")
47
- 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate
- Molecular Weight: g - Micelle Molecular Weight: 6149g - Critical Micelle Concentration (CMC): 8 to 10mM - Dialyzable: Yes
![- 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate](http://slideplayer.it/slide/2677197/10/images/47/-+3-%5B%283-Cholamidopropyl%29dimethylammonio%5D-1-propanesulfonate.jpg "- Molecular Weight: g. - Micelle Molecular Weight: 6149g. - Critical Micelle Concentration (CMC): 8 to 10mM. - Dialyzable: Yes.")
53
E’ importante lavorare a basse temperature
Un’analisi di sequenza della proteina target può evidenziare siti di taglio per specifiche proteasi E’ importante lavorare a basse temperature

54
Ottimizzazione dell’estratto
Il tampone di estrazione deve avere composizione salina e pH piu’ simile possibile alle condizioni cellulari, per mantenere in soluzione le proteine Forza ionica M pH 7-7.5 Generalmente 2-3 volumi di tampone rispetto al volume del pellet. Maggiore il volume minore sarà la % di proteine perse nel pellet Tipici tamponi utilizzati sono mM fosfato o Tris-HCl o HEPES Talvolta vengono aggiunti specifici stabilizzanti come, detergenti, EDTA, inibitori delle proteasi e reagenti contenenti gruppi sulfidrilici come -mercaptoetanolo e ditiotreitolo (5-20mM) Questi hanno lo scopo di proteggere i residui cisteinici dall’ossigeno I gruppi SH delle cisteine esposti all’ossigeno possono formare legami disolfuro –S-S- ossidarsi a gruppi solfinici –S-OH ossidarsi a gruppi solfonici La reazione 1) puo’ essere accelerata da ioni metallici. EDTA rimuove gli ioni metallici Reagenti con gruppi SH si ossidano all’ossigeno al posto delle cisteine
 Questi hanno lo scopo di proteggere i residui cisteinici dall’ossigeno. I gruppi SH delle cisteine esposti all’ossigeno possono. formare legami disolfuro –S-S- ossidarsi a gruppi solfinici –S-OH. ossidarsi a gruppi solfonici. La reazione 1) puo’ essere accelerata da ioni metallici. EDTA rimuove gli ioni metallici. Reagenti con gruppi SH si ossidano all’ossigeno al posto delle cisteine.")
55
Chiarificazione dell’estratto
L’estratto proteico, dopo la rottura delle cellule può essere viscoso per la presenza di acidi nucleici Questi possono essere rimossi: per aggiunta di RNAasi e DNAsi per precipitazione con solfato di protamina (composto policationico estratto da pesci) La protamina interagisce elettrostaticamente con gli acidi nucleici. Si forma un complesso che precipita a f. ionica non troppo elevata (< 0.1 M) e ne provoca la precipitazione L’estratto viene chiarificato da organelli, frammenti di membrana e precipitati per centrifugazione.
 La protamina interagisce elettrostaticamente con gli acidi nucleici. Si forma un complesso che precipita a f. ionica non troppo elevata (< 0.1 M) e ne provoca la. precipitazione. L’estratto viene chiarificato da organelli, frammenti di membrana e precipitati. per centrifugazione.")
56
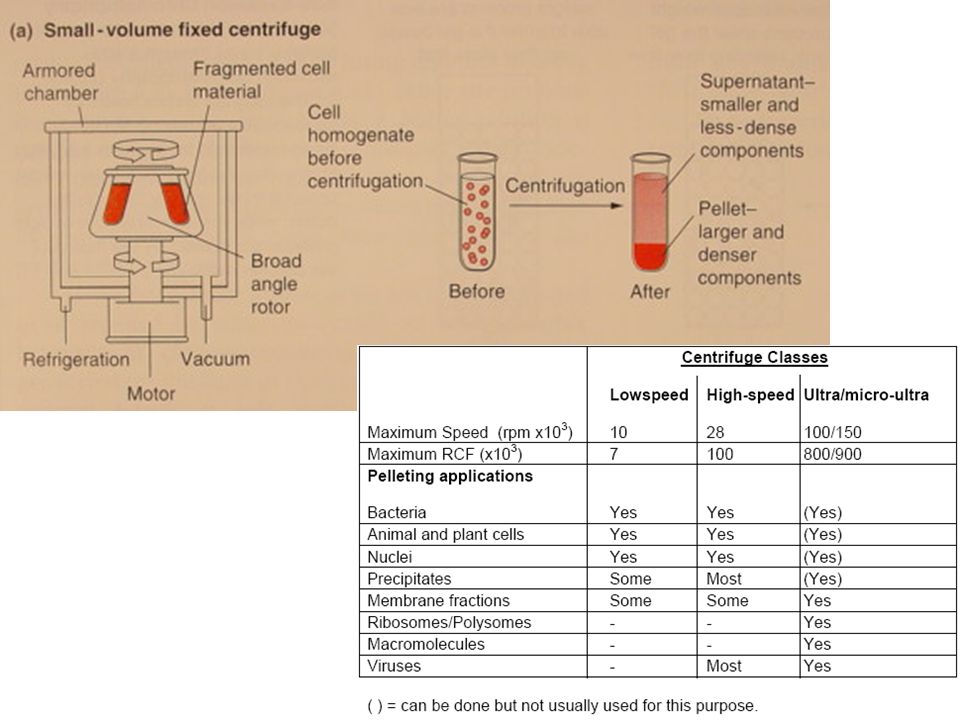
Differenza tra la sua densità e quella del mezzo
CHIARIFICAZIONE: CENTRIFUGAZIONE La velocità e la durata della centrifugazione dipendono dal coefficiente di sedimentazione (S) delle particelle da separare: Il coefficiente di sedimentazione è un numero adimensionale che misura il rapporto tra la velocità di sedimentazione di un corpo ideale (sfera) e quella del corpo in esame, a parità di condizioni di riferimento. In alcuni casi è espresso come unità di misura non SI denominata Svedberg 1 Svedberg = 10–13 sec S = 1/ω2r x dr/dt S enzimi, ormoni peptidici, proteine solubili 2-25 s Acidi nucleici 3-100 s Ribosomi e polisomi s virus s lisosomi 4.000 s membrane x103 s mitocondri 20x103S-70x103 s nuclei 4.000x103 S x103 s ω = velocità angolare [rad sec-1] = x RPM (rotazioni per min) r = distanza tra la particella ed il centro del rotore [mm] dr/dt = velocità di sedimentazione della particella [cm sec-1] dr/dt = [2/9 r2 p (p-m) 2 r]/ Differenza tra la sua densità e quella del mezzo Coefficiente di viscosità del mezzo Dimensione della particella
 delle particelle da separare: Il coefficiente di sedimentazione è un numero adimensionale che misura il rapporto tra la velocità di sedimentazione di un corpo ideale (sfera) e quella del corpo in esame, a parità di condizioni di riferimento. In alcuni casi è espresso come unità di misura non SI denominata Svedberg. 1 Svedberg = 10–13 sec. S = 1/ω2r x dr/dt. S. enzimi, ormoni peptidici, proteine solubili s. Acidi nucleici s. Ribosomi e polisomi s. virus s. lisosomi s. membrane x103 s. mitocondri. 20x103S-70x103 s. nuclei x103 S x103 s. ω = velocità angolare [rad sec-1] = x RPM (rotazioni per min) r = distanza tra la particella ed il centro del rotore [mm] dr/dt = velocità di sedimentazione della particella [cm sec-1] dr/dt = [2/9 r2 p (p-m) 2 r]/ Differenza tra la sua densità e quella del mezzo. Coefficiente di viscosità del mezzo. Dimensione della particella.")
58
CHIARIFICAZIONE: ULTRAFILTRAZIONE
Separazione tramite filtrazione con membrane a diverso CUT-OFF

59
ESTRAZIONE DI PROTEINE ESPRESSE IN CORPI DI INCLUSIONE

60
REFOLDING: L’agente denaturante viene rimosso per permettere la rinaturazione della proteina Tecniche di refolding: REFOLDING DIRETTO DIALISI REFOLDING IN COLONNA (proteine di fusione) Metodo sperimentale proprietà chimico-fisiche della proteina
 Metodo sperimentale proprietà chimico-fisiche della proteina.")
61
Preparazione materiale e soluzioni
¨ Sterilizzare materiale (punte, eppendorf, tubi da PCR) ¨ Sterilizzare 4 beute da 2 l, con 0.5 l ciascuna di 2YT ¨ Sterilizzare H2O ¨ Preparare 2 0.5 l 2YT sterile ¨ Preparare 2 10 ml amp sterile 1000 (2g/10 ml) ¨ Preparare 2 5 ml IPTG sterile (0.5 M) ¨ Preparare 2 10 piastre 2YT agar + amp (200 g/ ml) Soluzione sterile di ampicillina (2 g/10 ml) Sciogliere 2 g di ampicillina in 10 ml di H2O Sterilizzare per filtrazione in Falcon sterile Soluzione sterile di IPTG Sciogliere la quantità opportuna di IPTG, per avere soluzione 0.5 M, in 5 ml di H2O. Sterilizzare per filtrazione in Falcon sterile
 ¨ Sterilizzare 4 beute da 2 l, con 0.5 l ciascuna di 2YT. ¨ Sterilizzare H2O. ¨ Preparare 2 0.5 l 2YT sterile. ¨ Preparare 2 10 ml amp sterile 1000 (2g/10 ml) ¨ Preparare 2 5 ml IPTG sterile (0.5 M) ¨ Preparare 2 10 piastre 2YT agar + amp (200 g/ ml) Soluzione sterile di ampicillina (2 g/10 ml) Sciogliere 2 g di ampicillina in 10 ml di H2O. Sterilizzare per filtrazione in Falcon sterile. Soluzione sterile di IPTG. Sciogliere la quantità opportuna di IPTG, per avere soluzione 0.5 M, in 5 ml di H2O. Sterilizzare per filtrazione in Falcon sterile.")
62
Preparazione materiale e soluzioni
2YT l triptone g estratto di lievito g NaCl g Portare a volume con H2O – Sterilizzare in autoclave, 120 °C, 1 bar, 30’. 2YT agar l triptone g estratto di lievito g NaCl g Agar-agar g Aggiungere soluzione ampicillina sterile (concentrazione finale 200 g/ ml, 0.5 ml su 0.5 l) quando la soluzione e’ quasi fredda, prima che solidifichi
 quando la soluzione e’ quasi fredda, prima che solidifichi.")
Presentazioni simili




















 + soluto In genere solvente liquido (es.acqua) E soluto solido, liquido, aeriforme.>")

1 VELOCITA DI REAZIONE ED EQUILIBRI.>")










