Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
più generazioni interessate
tramissione da parte di entrambi i sessi circa il 50% dei soggetti affetti maschi e femmine affetti non segni di consanguineità Ereditarietà autosomica dominante

2
Trasmissione ereditaria dei caratteri mendeliani
Dominante - ogni tratto o carattere che si esprima nell’ eterozigote - nel caso di malattia: una sola copia del gene mutato è sufficiente per esprimere il fenotipo affetto Genotipo : eterozigote Fenotipo : affetto Codominante: l’eterozigote esprime un fenotipo distinto da quello dei due stati omozigoti per es. gruppi sanguigni, enzimi eritrocitari, etc.

3
Eredità autosomica dominante
Se un genitore è normale (bb) e l’altro è affetto (Bb) da una malattia autosomica dominante, il 50% dei figli sarà eterozigote affetto (Bb), ed il 50% sarà omozigote normale (bb). Nelle malattie autosomiche dominanti, la lettera maiuscola indica il gene che dà la malattia: un solo allele mutato dà origine al fenotipo affetto Rischio di ricorrenza = 50% (0.5)
 e l’altro è affetto (Bb) da una malattia autosomica dominante, il 50% dei figli sarà eterozigote affetto (Bb), ed il 50% sarà omozigote normale (bb). Nelle malattie autosomiche dominanti, la lettera maiuscola indica. il gene che dà la malattia: un solo allele mutato dà origine al fenotipo affetto. Rischio di ricorrenza = 50% (0.5)")
4
Eredità autosomica dominante
Se entrambi i genitori sono affetti (Bb) da una malattia autosomica dominante, allora il 75% dei figli sarà affetto (BB o Bb), e il 25% sarà omozigote normale (bb). Nota: è raro che un individuo affetto da una malattia autosomica dominante sia omozigote per il gene che dà la malattia.
 da una malattia autosomica dominante, allora il 75% dei figli sarà affetto (BB o Bb), e il 25% sarà omozigote normale (bb). Nota: è raro che un individuo affetto da una malattia autosomica. dominante sia omozigote per il gene che dà la malattia.")
5
Eredità autosomica dominante
si manifesta in uguale misura nei due sessi la trasmissione non dipende dal sesso il figlio di un genitore affetto e di uno sano ha il 50% di probabilità di essere affetto un individuo affetto ha comunemente un genitore affetto …………….mutazioni de novo individui non affetti non trasmettono la malattia ….……penetranza incompleta

6
a penetranza incompleta (penetranza del 70%) R (III-1) = 0.5 ???
Carattere AD a penetranza completa tasso m di “ nuove mutazioni ” R = 0.5 (50%) Carattere AD a penetranza incompleta (penetranza del 70%) R (III-1) = ??? R (IV-1) = 0.25 ???
 Carattere AD. a penetranza incompleta. (penetranza del 70%) R (III-1) = 0.5 R (IV-1) = 0.25")
7
Rischio di ricorrenza III-1
Teorema di Bayes: Rischio di ricorrenza III-1 Ipotesi Ipotesi 2 Portatore Non Portatore 1 / / prior p. 3 / conditional p. ½ x 3/10= 3/ ½ x 1=1/ joint p. 3/20 1/2 3/20 + 1/ /2 + 3/20 posterior p. = 3/13 (23%) = 10/13 (77%) R (III-1) di trasmettere = ½ x 3/13 = 3/26 (11.5%) R (III-1) di avere un figlio affetto = 3/26 x 7/10 = 8%
 = 10/13 (77%) R (III-1) di trasmettere = ½ x 3/13 = 3/26 (11.5%) R (III-1) di avere un figlio affetto = 3/26 x 7/10 = 8%")
8
Mutazioni nuove o “de novo”
identificate nelle malattie dominanti o recessive X-linked, molto raramente producono malattie autosomiche recessive (entrambe le copie del gene devono essere mutate) il rischio di ricorrenza è basso ma leggermente superiore all’incidenza della malattia nella popolazione a causa del mosaicismo della linea germinale oogenesi spermatogenesi Nuova mutazione mosaicismo
 il rischio di ricorrenza è basso. ma leggermente superiore all’incidenza della malattia nella. popolazione a causa del mosaicismo della linea germinale. oogenesi. spermatogenesi. Nuova mutazione. mosaicismo.")
9
Aspetti da considerare nelle malattie AD
Penetranza - completa – incompleta - diversa per fasce di età Espressività - presenza – assenza - espressività variabile Tasso di nuove mutazioni casi sporadici – famigliari Percentuale di mosaicismi germinali

10
Retinoblastoma (1:15.000-20.000) 60% dei casi 40% dei casi
penetranza >90% Ipotesi: mosaicismo germinale o somatico Rb espressività variabile Rb Rb Rb bilaterale Rb multifocale monolaterale Retinoblastoma sporadico monolaterale unico

11
Retinoblastoma f 1 : 15.000-20.000 nati
60% dei casi: tumore monolaterale unico inattivazione somatica di Rb1 (-/-) 40% dei casi: tumore multifocale e/o bilaterale con storia famigliare POSITIVA trasmissione AD di mutazione Rb1 (-/+) : inattivazione somatica del secondo allele (-/-) con storia famigliare NEGATIVA mutazione de novo in un gamete (maschile) mosaicismo germinale in un genitore (6-10%) mosaicismo somatico nell’affetto (10%) RR=0.5, RT=0.5 RR=??, RT=??
 40% dei casi: tumore multifocale e/o bilaterale. con storia famigliare POSITIVA. trasmissione AD di mutazione Rb1 (-/+) : inattivazione somatica del secondo allele (-/-) con storia famigliare NEGATIVA. mutazione de novo in un gamete (maschile) mosaicismo germinale in un genitore (6-10%) mosaicismo somatico nell’affetto (10%) RR=0.5, RT=0.5. RR= , RT=")
12
Gene Rb1 13q14: 27 esoni, mRNA 4,840 , proteina 928 aa
più di 400 mutazioni note 85-90% mutazioni troncanti (NS, FS, SS): esoni 2-25
: esoni")
13
che tipo di ereditarietà
SCA : che tipo di ereditarietà qual’è la probabilità di essere portatore per il padre e per la probanda è possibile eseguire un test genetico ? A che scopo ? a chi offrirlo

14
che tipo di ereditarietà Autosomica Dominante
SCA : che tipo di ereditarietà qual’è la probabilità di essere portatore per il padre e per la probanda è possibile eseguire un test genetico ? A che scopo ? a chi offrirlo Autosomica Dominante Ipotesi 1 Ipotesi 2 Portatore Non Portatore 1 / / prior p. 2 / condit. p. ½ x 2/10= 2/20 ½ x 1=1/2 joint p. 1/ /2 1/10 + 1/ /2 + 1/10 post. p. = 1/6 (16.6%) = 5/6 (83.3%) Test ai membri affetti conferma della diagnosi esclusione certa dei non portatori definizione del RR della probanda Probanda: 1/6 x ½ = 1/12 (8.3%)
 = 5/6 (83.3%) Test ai membri affetti. conferma della diagnosi. esclusione certa dei non. portatori. definizione del RR della. probanda. Probanda: 1/6 x ½ = 1/12 (8.3%)")
15
SCA1 SCA2 SCA3 SCA6 SCA7 SCA8 SCA10 SCA12 SCA17 Malattia (9 forme)
Locus Ripetizioni normali Ripetizioni patologiche Frequenza tra le ADCA (Italia) Caratteristiche distintive segni piramidali, neuropatia periferica SCA1 6p23 CAG/6-44 39-83 21% SCA2 movimenti saccadici lenti, neuropatia periferica 12q24.1 CAG/14-31 36-400 23% nistagmo, retrazione delle palpebre, fascicolazioni amiotrofiche SCA3 14q21 CAG/13-43 55-86 <1% atassia episodica, decorso molto lento SCA6 19p13.1 CAG/ 4-18 (19)20-33 <1% perdita della vista con neuropatia SCA7 3p21.1-p12 CAG / 4-19 37->300 <1% DTR vivaci e diminuzione della sensibilità vibratoria SCA8 13q21 CTG/ 15-50 80-250 <1% attacchi epilettici occasionali SCA10 22q13 ATTCT/ 10-22 assente tremori precoci, demenza tardiva SCA12 5q31 CAG / 6-26 66-78 assente SCA17 6q27 CAG/ 30-42 45-63 <1% deterioramento mentale
 Caratteristiche. distintive. segni piramidali, neuropatia periferica. SCA1. 6p23. CAG/ % SCA2. movimenti saccadici. lenti, neuropatia periferica. 12q24.1. CAG/ % nistagmo, retrazione delle palpebre, fascicolazioni amiotrofiche. SCA3. 14q21. CAG/ <1% atassia episodica, decorso molto lento. SCA6. 19p13.1. CAG/ (19) <1% perdita della vista. con neuropatia. SCA7. 3p21.1-p12. CAG / >300. <1% DTR vivaci e diminuzione. della sensibilità vibratoria. SCA8. 13q21. CTG/ <1% attacchi epilettici. occasionali. SCA10. 22q13. ATTCT/ assente. tremori precoci, demenza tardiva. SCA12. 5q31. CAG / assente. SCA17. 6q27. CAG/ <1% deterioramento mentale.")
16
SCA : 13 loci noti SCA4 SCA5 SCA9 SCA11 SCA13 SCA14 SCA15 SCA16 SCA18
Malattia Locus N° di famiglie descritte Caratteristiche distintive SCA4 16q22.1 >5 (japan/scand.) neuropatia assonale sensitiva SCA5 11p12 1 (Lincoln) insorgenza precoce, decorso lento SCA9 X (riservato) SCA11 15q14 -q21 2 (U.K.) lieve, rimane la deambulazione SCA13 19q13 1 (french) lieve ritardo mentale, bassa statura Yellow: loci with published markers SCA14 19q13.4 2 (japan/english) mioclono assiale precoce SCA15 3pter-24.2 1 (australian) atassia pura e progressione lenta SCA16 8q22-24 1 (japan) neuropatia sens., atrofia musc. SCA18 7q31-32 ? tremore della testa SCA19 1p21-q21 1 (dutch) lieve atassia, segni piram., dist. cogn. SCA20 riservato SCA21 7p21.3-p15.1 1 (french) progressione lenta, rigidità, tremore SCA22 1q22 (riservato)
 neuropatia assonale sensitiva. SCA5. 11p12. 1 (Lincoln) insorgenza precoce, decorso lento. SCA9. X (riservato) SCA11. 15q14 -q21. 2 (U.K.) lieve, rimane la deambulazione. SCA13. 19q13. 1 (french) lieve ritardo mentale, bassa statura. Yellow: loci with published markers. SCA14. 19q (japan/english) mioclono assiale precoce. SCA15. 3pter (australian) atassia pura e progressione lenta. SCA16. 8q (japan) neuropatia sens., atrofia musc. SCA18. 7q tremore della testa. SCA19. 1p21-q21. 1 (dutch) lieve atassia, segni piram., dist. cogn. SCA20. riservato. SCA21. 7p21.3-p (french) progressione lenta, rigidità, tremore. SCA22. 1q22 (riservato)")
17
una sola generazione interessata
ramo paterno a materno sani maschi e femmine affetti talora segni di consanguineità Ereditarietà autosomica recessiva

18
ogni tratto o carattere che si esprima solo nell’ omozigote
Trasmissione ereditaria dei caratteri mendeliani Recessiva : ogni tratto o carattere che si esprima solo nell’ omozigote nel caso di malattia: entrambe le copie del gene mutato devono essere presenti per esprimere un fenotipo affetto Genotipo : omozigote Fenotipo : affetto Genotipo : eterozigote Fenotipo : portatore sano I fenotipi possono essere analizzati e misurati a differenti livelli. Es. malattie metaboliche: gli eterozigoti sono sani ma l’enzima responsabile della malattia può avere nel siero una concentrazione intermedia rispetto a quella dei due stati omozigoti.

19
a Eredità autosomica recessiva
Se un genitore è normale (AA) e l’altro è portatore (Aa), allora il 50% in media dei figli sarà omozigote normale (AA), e l’altro 50% sarà portatore eterozigote non affetto (Aa). Non vi sono figli affetti. Nel caso di malattie autosomiche recessive, le lettere minuscole stanno ad indicare i geni che in omozigosi danno la malattia, perché entrambi gli alleli devono essere mutati perché si osservi il fenotipo patologico. Qual’è la probabilità di avere un figlio affetto se entrambi i genitori sono portatori?
 e l’altro è portatore (Aa), allora il 50% in media dei figli sarà omozigote normale (AA), e l’altro 50% sarà portatore eterozigote non affetto (Aa). Non vi sono figli affetti. Nel caso di malattie autosomiche recessive, le lettere minuscole stanno ad indicare i geni che in omozigosi danno la malattia, perché entrambi gli alleli devono essere mutati perché si osservi il fenotipo patologico. Qual’è la probabilità di avere un figlio affetto se entrambi i genitori sono portatori")
20
Eredità autosomica recessiva
Se entrambi i genitori sono portatori sani (Aa), in media il 25% dei figli sarà omozigote normale (AA), il 50% sarà eterozigote non affetto (Aa), e il 25% sarà omozigote affetto (aa). Se un genitore è un portatore sano (Aa) e l’altro è affetto (aa), allora il 50% in media dei figli sarà eterozigote non affetto (Aa), e l’altro 50% sarà omozigote affetto (aa).
, in media il 25% dei figli sarà omozigote normale (AA), il 50% sarà eterozigote non affetto (Aa), e il 25% sarà omozigote affetto (aa). Se un genitore è un portatore sano (Aa) e l’altro è affetto (aa), allora il 50% in media dei figli sarà eterozigote non affetto (Aa), e l’altro 50% sarà omozigote affetto (aa).")
21
Eredità autosomica recessiva
• gli individui affetti sono omozigoti (o eterozigoti composti) • nella maggior parte dei casi, entrambi i genitori sono portatori sani – in media, 1 su 4 figli è affetto • la trasmissione non dipende dal sesso • matrimoni tra individui affetti e non affetti genera solo figli eterozigoti sani – a meno che il partner non affetto sia eterozigote • più è rara la malattia, più è probabile che l’individuo affetto sia figlio di genitori tra loro consanguinei (inbreeding)
 • nella maggior parte dei casi, entrambi i genitori sono portatori sani. – in media, 1 su 4 figli è affetto. • la trasmissione non dipende dal sesso. • matrimoni tra individui affetti e non affetti genera solo figli eterozigoti sani. – a meno che il partner non affetto sia eterozigote. • più è rara la malattia, più è probabile che l’individuo affetto sia figlio di genitori tra loro consanguinei (inbreeding)")
22
½ x f x ¼ AA+Aa Qual è la probabilità di avere un figlio affetto
? AA+Aa Qual è la probabilità di avere un figlio affetto per III-2 ? Probabilità di essere portatore: ½ Probabilità che la coniuge sia portatrice: f portatori nella popolazione ½ x f x ¼ III-2

23
Frequenza (f) dei portatori nella popolazione generale
Legge di Hardy-Weinberg p frequenza allele “normale” (A) q frequenza allele “mutato” (a) i genotipi nella popolazione saranno : p2 + q2 + 2pq = (p + q)2 dove p + q = 1 Conoscendo la frequenza degli affetti : q2 (aa) p = q2 2pq (f portatori) = 2 ( q ) q2
 q frequenza allele mutato (a) i genotipi nella popolazione saranno : p2 + q2 + 2pq = (p + q)2 dove p + q = 1. Conoscendo la frequenza degli affetti : q2 (aa) p = 1 - q2. 2pq (f portatori) = 2 (1 - q2 ) q2.")
24
Legge di Hardy-Weinberg p2 : q2 : 2pq
La distribuzione binomiale presume che : la popolazione sia ampia gli incroci siano casuali le frequenze alleliche siano costanti - frequenza di nuove mutazioni - fitness riproduttiva degli omozigoti affetti - “deriva genetica” - vantaggi degli eterozigoti - migrazioni

25
Qual è la probabilità di avere un figlio affetto per III-2 ? ½ x f x ¼ AA+Ay Ay Ax xy Se l’individuo IV-2 ha un genotipo noto: mutX/mutY ? -> testing genetico risolutivo Se III-2 è Ay = RR= 1 x f x ¼ Se III-2 è AA = RR pressochè nullo

26
Qual è la probabilità di avere un figlio affetto per III-2 ? ½ x f x ¼ A? Ax x? Se nell’individuo IV-2 il test genetico ha permesso di identificare una sola mutazione (mutX) ? Stato genetico di III-2 indeterminato RR= ½ x f x ¼ Se III-1 (coniuge) ha un test genetico negativo : RR = ½ x f (sensibilità del test) x ¼
 Stato genetico di III-2 indeterminato. RR= ½ x f x ¼. Se III-1 (coniuge) ha un test genetico. negativo : RR = ½ x f (sensibilità del test) x ¼.")
27
Dominanza o Recessività
Dipende da : caratteristiche della malattia ? caratteristiche del gene ? effetto biologico delle mutazioni ? effetto biologico delle mutazioni in relazione alle caratteristiche funzionali della proteina (prodotto genico)
")
28
Malattie ereditarie recessive
Mutazioni che inducono una perdita di funzione Gli eterozigoti (portatori) sono normali, una riduzione del prodotto proteico del 50% viene tollerato se il rimanente 50% è sufficiente per una funzione normale Esempio: tratto falcemico, bA bS Gli omozigoti sono affetti perché non viene prodotta proteina o quella che è prodotta non funziona normalmente Esempio: anemia falciforme, bS bS difetti enzimatici recessivi, fibrosi cistica
 sono normali, una riduzione del prodotto proteico del 50% viene tollerato se il rimanente 50% è sufficiente per una funzione normale. Esempio: tratto falcemico, bA bS. Gli omozigoti sono affetti perché non viene prodotta proteina o quella che è prodotta non funziona normalmente. Esempio: anemia falciforme, bS bS. difetti enzimatici recessivi, fibrosi cistica.")
29
Mutazioni delle “serpine” (inibitori delle serin-proteasi)
a1-antitripsina anti-elastasi dei neutrofili antitrombina anti-proteasi della coagulazione inibitore di C1 anti-attivazione del complemento inibitori della plasmina anti-fibrinolisi

30
Mutazioni dell’ a1-antitripsina (AR) (inibitore dell’elastasi dei neutrofili)
mutazioni “loss of function” : ins o del (frame shift) deficit di enzima circolante eccesso di attività dell’elastasi leucocitaria enfisema mutazioni missenso del ”sito reattivo” : Met358Arg conversione in antitrombina malattia emorragica fatale mutazioni missenso nella ”regione mobile o b sheet” : Glu342Lys polimerizzazione della serpina accumulo negli epatociti (ER) fibrosi portale cirrosi ed epatocarcinoma (colangiocarcinoma)
 deficit di enzima circolante. eccesso di attività dell’elastasi leucocitaria. enfisema. mutazioni missenso del sito reattivo : Met358Arg. conversione in antitrombina. malattia emorragica fatale. mutazioni missenso nella regione mobile o b sheet : Glu342Lys. polimerizzazione della serpina. accumulo negli epatociti (ER) fibrosi portale. cirrosi ed epatocarcinoma (colangiocarcinoma)")
31
Connessina 26 GJB2 (Gap Junction Beta 2)
proteina di 226 aa esamero (connessone): canale del potassio espressione: stria vascolare, legamento spirale, tra le cellule di sostegno nella coclea, cute elevato contenuto di K+ nell’endolinfa
: canale del potassio. espressione: stria vascolare, legamento spirale, tra le cellule di sostegno nella coclea, cute. elevato contenuto di K+ nell’endolinfa.")
32
Connessina 26 GJB2 (Gap Junction Beta 2)
frameshift e stop 35DG In Italia : in omozigosi o in eterozigosi composta nel 40% circa di tutti i casi di sordità congenita non sindromica recessiva

33
Connessina 26 GJB2 (Gap Junction Beta 2)
C202F W44C Alleli dominanti : sordità post-linguale progressiva AD

34
Connessina 26 GJB2 (Gap Junction Beta 2)
D66H R75W Alleli dominanti : Sindrome di Vohwinkel : sordità congenita AD ipercheratosi palmo-plantare

35
Fibrosi cistica (AR, 1/2500) malattia cronica polmonare
ostruzione diffusa delle vie aeree di piccolo calibro infezioni polmonari ricorrenti, fibrosi, cisti, ascessi anomalie gastrointestinali e malassorbimento Ileo da meconio (15-20%) insufficienza pancreatica, pancreatite cronica (92%) malattia epato-biliare cronica (6%) ostruzione intestinale, prolasso rettale malassorbimento (vit. liposolubili), ritardo di crescita azoospermia ostruttiva assenza dei vasi deferenti azoospermia, ridotto liquido seminale, pH<7 sindromi da perdita di Sali sopravvivenza: 31.6 aa (56aa ps – 20-25aa pi)
 insufficienza pancreatica, pancreatite cronica (92%) malattia epato-biliare cronica (6%) ostruzione intestinale, prolasso rettale. malassorbimento (vit. liposolubili), ritardo di crescita. azoospermia ostruttiva. assenza dei vasi deferenti. azoospermia, ridotto liquido seminale, pH<7. sindromi da perdita di Sali. sopravvivenza: 31.6 aa (56aa ps – 20-25aa pi)")
36
Fibrosi cistica (AR, 1/2500) Tripsinogeno immunoreattivo
screening neonatale Test del sudore Cl > 60mEq/L in due separati dosaggi in una quantità di almeno 75mg (30 minuti) Potenziale nasale di trans-membrana dopo i sei anni (potenziale più negativo) Analisi genetica per la ricerca di mutazioni “frequenti” Ricerca di mutazioni di secondo livello
 Potenziale nasale di trans-membrana. dopo i sei anni (potenziale più negativo) Analisi genetica per la ricerca di mutazioni frequenti Ricerca di mutazioni di secondo livello.")
37
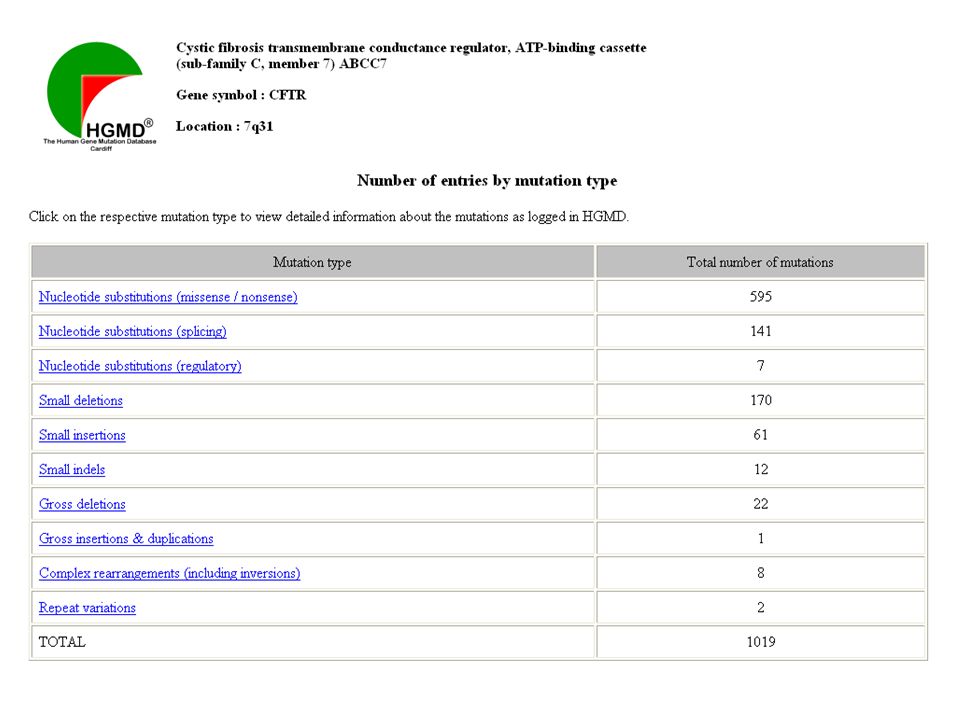
Fibrosi cistica (AR, 1/2500) 7q31.2 gene CFTR
27 esoni mRNA: 6128 b aa

38
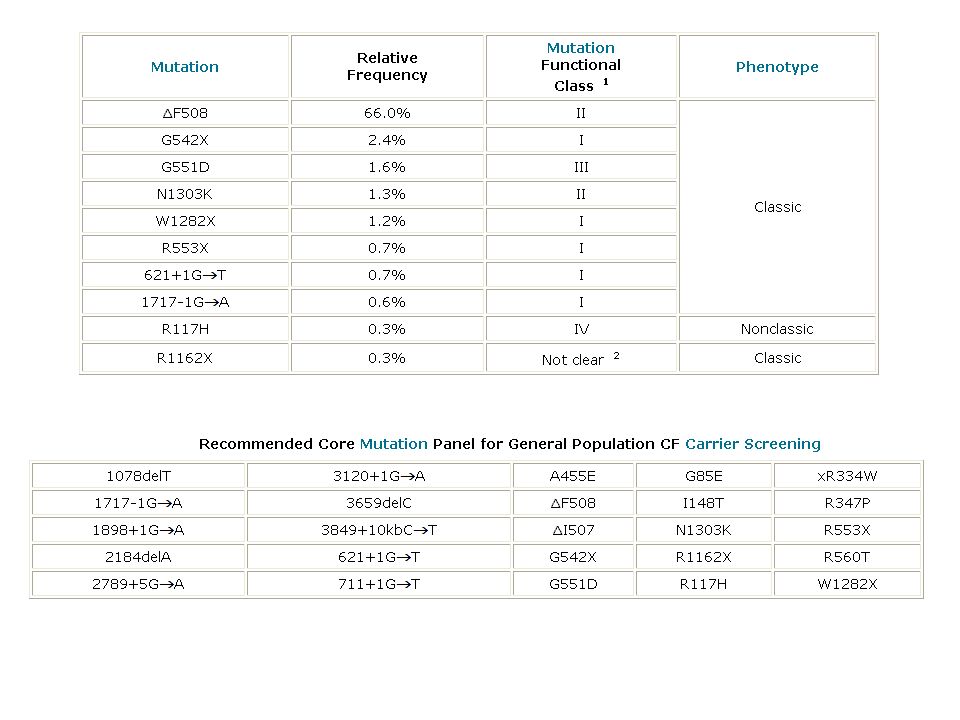
Mutazioni CFTR e Fibrosi cistica
230 kb 27 esoni 1480 aa proteina ABC ATP-binding cassette

39
Mutazioni CFTR e Fibrosi cistica

43
Test OLA (31-33 mutazioni): sensibilità del 75% 1/28 27/28 1/4 1
f = 1/28 RA = 1/28 • 1/28 • 1/4 = 1/3100 ~ PORTATORE NON PORTATORE 1/28 27/28 probabilità a priori 1/4 1 test OLA negativo 1/28 • 1/4 = 1/112 27/28 • 1 = 27/28 Test OLA negativo 3/4 P mut+ (75%) 1/4 P mut- (25%) 1/112 1/ /28 = 1/109 RA = 1/109 • 1/109 • 1/4 = 1/47500 ~
 1/4 P. mut- (25%) 1/112. 1/ /28. = 1/109. RA = 1/109 • 1/109 • 1/4 = 1/47500 ~")
44
DF508 e test OLA Oligonucleotide Ligation Assay
CFTR cDNA Esone 10 1561 CCTTCAGAGGGTAAAATTAAGCACAGTGGAAGAATTTCATTCTGTTCTCAGTTTTCCTGG 477 -P--S--E--G--K--I--K--H--S--G--R--I--S--F--C--S--Q--F--S--W- 1621 ATTATGCCTGGCACCATTAAAGAAAATATCATCTTTGGTGTTTCCTATGATGAATATAGA 497 -I--M--P--G--T--I--K--E--N--I--I--F--G--V--S--Y--D--E--Y--R- 1681 TACAGAAGCGTCATCAAAGCATGCCAACTAGAAGAGGACATCTCCAAGTTTGCAGAGAAA 517 -Y--R--S--V--I--K--A--C--Q--L--E--E--D--I--S--K--F--A--E--K- 1741 GACAATATAGTTCTTGGAGAAGGTGGAATCACACTGAGTGGAGGTCAACGAGCAAGAATT 537 -D--N--I--V--L--G--E--G--G--I--T--L--S--G--G--Q--R--A--R--I- Esone 11 TTTCTTTTATAGTAGAA ATTATGCCTGGCACCATTAAAGAAAATATCATCTTTGGTGTTTCCTATGATGAATATAGA XXXXXXXXXXXXXXXXXXX TAATTTCTTTTATAGTA ATTATGCCTGGCACCATTAAAGAAAATATCATTGGTGTTTCCTATGATGAATATAGA XXXXXXXXXXXXXXX Seq. wt Seq. mut

45
Reazione di Ligasi e amplificazione mediante PCR
TTTCTTTTATAGTAGAAACCACAAAGGATACTACTT ATTATGCCTGGCACCATTAAAGAAAATATCATCTTTGGTGTTTCCTATGATGAATATAGA XXXXXXXXXXXXXXXXXXX TAATTTCTTTTATAGTAACCACAAAGGATACTACTT ATTATGCCTGGCACCATTAAAGAAAATATCATTGGTGTTTCCTATGATGAATATAGA XXXXXXXXXXXXXXX XXXXXXXXXXXXXXXXXXX Reazione di Ligasi e amplificazione mediante PCR con primers specifici per le code: Pannello di prodotti di PCR di dimensioni diverse a seconda della specificità dell’oligonucleotide separati con elettroforesi capillare Es. F508 wt : 74 bp DF508 : bp

46
1986: diagnosi di azoospermia associata ad assenza dei vasi deferenti.
1958 1965 1986: diagnosi di azoospermia associata ad assenza dei vasi deferenti. Prelievo di spermatozoi e tentativi di fecondazione assistita falliti….in attesa di ICSI. Marito: CFTR D1270N-5T introne 8 Moglie: CFTR negativo per le 31 mutazioni più frequenti e analisi di 10 esoni in DGGE rischio residuo di essere portatrice: 1/200 Rischio per la coppia di avere un figlio affetto da una forma lieve di fibrosi cistica o CBAVD di 1/400

47
Mutazioni CFTR e CBAVD Sono spesso presenti (70%) segni sub-clinici di fibrosi cistica: test al sudore alterato (> 40 mEqu/l) ostruzione nasale, polipi nasali, sinusiti infezioni respiratorie ricorrenti

48
Mutazioni dominanti Mutazioni dominanti negative
proteina che non funziona e che inibisce o interferisce con la funzione della proteina normale codificata dall’allele sano tipico delle proteine multimeriche

49
Osteogenesi imperfetta (AD)
Collagene tipo I 2 molecole di Col a 1 (17q) 1 molecola di Col a 2 (7q) (338 ripetizioni GXY)
 1 molecola di Col a 2 (7q) (338 ripetizioni GXY)")
50
Osteogenesi imperfetta (AD)
OI lieve (tipo I) statura normale, scarse deformità sclere blu e sordità, fratture OI letale-grave (tipo II-III) fratture multiple accorciamento delle ossa lunghe deformità ossee, sordità mut de novo in Gly 5-10% : mosaicismo germinale OI moderata (tipo IV) bassa statura, deformità osteoporosi, dentinogenesi imperfetta
 statura normale, scarse deformità. sclere blu e sordità, fratture. OI letale-grave (tipo II-III) fratture multiple. accorciamento delle ossa lunghe. deformità ossee, sordità. mut de novo in Gly. 5-10% : mosaicismo germinale. OI moderata (tipo IV) bassa statura, deformità. osteoporosi, dentinogenesi imperfetta.")
51
Mutazioni dominanti Mutazioni che inducono un eccesso di funzione :
proteina con funzione o espressione alterata es. malattia di Charcot-Marie-Tooth (17p11.2): duplicazione di 1.5 Mb di DNA (3 copie) sovraesprime la proteina della mielina periferica (PMP22) es. neoplasie endocrine multiple tipo 2 (10q11.2): mutazioni missenso del recettore RET con attivazione costitutiva ligando indipendente
: duplicazione di 1.5 Mb di DNA (3 copie) sovraesprime la proteina della mielina periferica (PMP22) es. neoplasie endocrine multiple tipo 2 (10q11.2): mutazioni missenso del recettore RET con attivazione costitutiva ligando indipendente.")
52
Recettore III per fattore di crescita dei fibroblasti
Mutazioni nel FGFR3 Recettore III per fattore di crescita dei fibroblasti Fenotipi: Acondroplasia Ipocondroplasia Displasia tanatofora Acondroplasia Severa con ritardo di sviluppo e acantosi nigricans

53
Mutazioni dominanti Insufficienza del corredo aploide (aploinsufficienza) : la perdita di funzione di una copia del gene (50%) comporta una riduzione critica della proteina es. ipercolesterolemia: mutazioni nel recettore della lipoproteina a bassa densità (LDL) con diminuzione dei livelli di recettore ->circa il doppio di colesterolo rimane in circolo es. mutazioni RET nella malattia di Hirchsprung es. delezioni del locus PMP22 nella neuropatia tomaculare
 comporta una riduzione critica della proteina. es. ipercolesterolemia: mutazioni nel recettore della lipoproteina a bassa densità (LDL) con diminuzione dei livelli di recettore. ->circa il doppio di colesterolo rimane in circolo. es. mutazioni RET nella malattia di Hirchsprung. es. delezioni del locus PMP22 nella neuropatia tomaculare.")
54
Mutazioni dominanti Proteine “ tossiche “ per alcuni tessuti :
L’espansione di triplette CAG nella regione codificante di alcuni geni producono lunghi tratti di poli-Glu es. Corea di Huntington e Atassie spino-cerelellari: proteine con poli-Glu che sono tossiche per le cellule nervose dei nuclei della base con formazione di inclusioni nucleari e morte cellulare

55
Malattie associate ad espansioni di triplette ripetute
Le espansioni ripetute di trinucleotidi interferiscono con l’espressione del gene o della proteina codificata

56
Mutazioni dominanti effetto “tossico” dell’RNA messaggero Gene FMR1
CGG: POF FXTAS CGG > 200 - FMR

57
Mutazioni dominanti Mutazioni germinali dominanti ma “recessive a livello cellulare” : Mutazioni che causano perdita di funzione di un allele conferiscono un “rischio di malattia”. Le cellule “patologiche” perdono per mutazione somatica l’allele WT es. Suscettibilità al cancro da mutazioni in geni onco-soppressori.

58
Inattivazione di geni onco - soppressori : Inattivazione di un allele
perdita di funzione Inattivazione di un allele Inattivazione del secondo allele per mutazione o delezione Both alleles of the tumor suppressor gene must be inactivated for a tumor to form. In sporadic tumors one allele is occasionaly inactivated and a second mutation of the funcional allele give the total inactivation of the tumor suppressor gene. In familial cancer predisposition syndromes, a mutant allele is inherited and is present in every cell. But the neoplastic fenotype arise only when the second allele is inactivated. (10-10 x bp x mitosi in 6x1012 bp di genoma)
")
59
Gene onco-soppressore Rb1
Alleli Rb normali Mutazione germinale mutazione somatica mutazione somatica mutazione somatica Retinoblastomi multipli bilaterali ad insorgenza precoce Retinoblastoma unico mono-laterale

60
Inattivazione di geni onco - soppressori
con perdita di eterozigosità (LOH) Allele 1 5 8 Allele 2 3 6 Prima mutazione: puntiforme Allele 1 5 8 Allele 2 3 6 seconda mutazione: delezione Allele 8 Allele 2 3 6
 Allele Allele Prima mutazione: puntiforme. Allele Allele seconda mutazione: delezione. Allele. 8. Allele")
61
Inattivazione di geni onco - soppressori
con perdita di eterozigosità (LOH) Allele 3 1 5 8 Allele 3 2 6 Allele 1 4 6 8 Allele 3 2 6 Allele 5 3 4 6 Allele 3 2 6 Tessuto sano Allele 8 Allele 3 2 6 Allele 1 8 Allele 3 2 6 Allele 5 3 Allele 3 2 6 Tessuto tumorale Loci studiati: Caso 1 ni - - + Caso Caso ni Sede del gene onco-soppressore
 Allele Allele Allele Allele Allele Allele Tessuto. sano. Allele. 8. Allele Allele Allele Allele Allele Tessuto. tumorale. Loci studiati: Caso 1 ni Caso Caso ni. Sede del gene onco-soppressore.")
62
TTGCCGGCCCCACAACCCTTG
SDHB exon 1 45_46insCC TTGCCGGCCCCACAACCCTTG D1S3669 D1S170 PBL GIST PGL 436 kb centr 50 kb telom

Presentazioni simili


 Dominanza incompleta>")

 è dominante sul pelo liscio ( r )>")










 DI BREVI TRATTI RIPETUTI>")




