Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
CLONAZIONE MOLECOLARE Corso di Biotecnologie
LA TECNOLOGIA del DNA RICOMBINANTE o CLONAZIONE MOLECOLARE Corso di Biotecnologie 12 Maggio 2008

2
Rappresentazione schematica del clonaggio (I)
DNA SORGENTE VETTORE DI CLONAZIONE 1 DNA BERSAGLIO frammentazione enzimatica linearizzazione enzimatica 2 3 congiungimento DNA bersaglio- vettore di clonazione

3
introduzione del DNA nella
cellula ospite (trasformazione) e isolamento delle cellule con il gene clonato cellula ospite proteina codificata dal gene clonato Produzione della proteina tutto ciò è possibile… grazie alla scoperta delle endonucleasi di restrizione
 e isolamento delle cellule con il gene clonato. cellula ospite. proteina codificata. dal gene clonato. Produzione della proteina. tutto ciò è possibile… grazie alla scoperta delle endonucleasi di restrizione.")
4
Le Endonucleasi di Restrizione (I)
Enzimi batterici: endonucleasi di TIPO II: Riconoscono e tagliano corte sequenze di DNA (4-8bp); le sequenze riconosciute sono palindromiche, cioè risultano ugluali se lette su ogni filamento nella stessa direzione il taglio produce estremità piatte o coesive Esempio di una delle prime endonucleasi di tipo II meglio caratterizzate: EcoRI genere: Escherichia Ordine di caratterizzazione di diverse endonucleasi appartenente allo stesso organismo (I) specie: Coli ceppo: R 5’- GAATTC - 3’ 3’- CTTAAG – 5’ SITO DI RICONOSCIMENTO EcoRI Il taglio genera 4 basi sporgenti in singolo filamento 5’- G AATTC - 3’ 3’- CTTAA G – 5’ OH P PRODOTTI FORMAZIONE DI ESTREMITÀ COESIVE
; le sequenze riconosciute sono palindromiche, cioè risultano ugluali se lette su ogni filamento nella stessa direzione. il taglio produce estremità piatte o coesive. Esempio di una delle prime endonucleasi di tipo II meglio caratterizzate: EcoRI. genere: Escherichia. Ordine di caratterizzazione di diverse endonucleasi appartenente allo stesso organismo (I) specie: Coli. ceppo: R. 5’- GAATTC - 3’ 3’- CTTAAG – 5’ SITO DI. RICONOSCIMENTO. EcoRI. Il taglio genera 4 basi sporgenti in singolo filamento. 5’- G AATTC - 3’ 3’- CTTAA G – 5’ OH. P. PRODOTTI. FORMAZIONE. DI ESTREMITÀ. COESIVE.")
5
Le Endonucleasi di Restrizione (II)
Enzima Sito di riconoscimento EcoR I G A A T T C C T T A A G BamH I G G A T C C C C T A G G Hind III A A G C T T T T C G A A KPN I G G T A C C C C A T G G NOT I G C G G C C G C C G C C G G C G EcoR V G A T A T C C T A T A G Xho I C T C G A G G A G C T C Isoschizomeri Sebbene gli enzimi di restrizione isolati siano oltre 3500, le sequenze bersaglio che possono essere tagliate sono molte di meno (appena più di duecento). E’ evidente che molti enzimi isolati da batteri diversi hanno la stessa specificità di sequenza,sono cioè isoschizomeri.
. E’ evidente che molti enzimi isolati da batteri diversi hanno la stessa specificità di sequenza,sono cioè isoschizomeri.")
6
…grazie alla scoperta delle endonucleasi di restrizione…
Le librerie geniche: collezione di cloni derivante dalla totalità di DNA estratto da un organismo e digerito con un enzima di restrizione: Le librerie genomiche: composta dalla totalità del DNA cromosomico di un organismo (es: se si vuole studiare l’organizzazione genica); Le librerie di cDNA: composta dall’mRNA di una cellula o di un tessuto in un particolare momento (es: studiare il profilo di espressione di una particolare proteina in u particolare momento o tessuto) Confronto tra I principali passaggi richiesti per la costruzione di librerie genomiche e di cDNA Libreria genomica 1 Libreria di cDNA Estrazione del DNA cromosomico 2 Estrazione dell’mRNA 2 Taglio del DNA con endonucleasi di restrizione (digestione parziale o totale) 3 Produzione di cDNA (trascrittasi inversa) 3 Trascrittasi inversa: isolata da retrovirus a RNA. Non si usa l’mRNA perchè è molto instabile. Per le librerie genomiche se si usa un enzima che rionosce sequenze tetranucleotidiche questo genererebbe frammenti troppo piccoli da poter poi clonare e quindi si fa una digestione parziale! Inserimento di ogni frammento di DNA in un vettore 4 Inserimento di ogni cDNA in un vettore 4 5 Trasformazione batterica
; Le librerie di cDNA: composta dall’mRNA di una cellula o di un tessuto in un particolare momento (es: studiare il profilo di espressione di una particolare proteina in u particolare momento o tessuto) Confronto tra I principali passaggi richiesti per la costruzione di librerie genomiche e di cDNA. Libreria genomica. 1. Libreria di cDNA. Estrazione del DNA. cromosomico. 2. Estrazione dell’mRNA. 2. Taglio del DNA con. endonucleasi di restrizione. (digestione parziale o totale) 3. Produzione di cDNA. (trascrittasi inversa) 3. Trascrittasi inversa: isolata da retrovirus a RNA. Non si usa l’mRNA perchè è molto instabile. Per le librerie genomiche se si usa un enzima che rionosce sequenze tetranucleotidiche questo genererebbe frammenti troppo piccoli da poter poi clonare e quindi si fa una digestione parziale! Inserimento di ogni frammento. di DNA in un vettore. 4. Inserimento di ogni cDNA. in un vettore Trasformazione batterica.")
7
Screening di librerie geniche
1. Ibridazione su colonia del DNA si piastrano cellule provenienti dalla reazione di trasformazione su terreno con antibiotico Piastra madre Trasferimento cellule ciascuna colonia formatasi sulla piastra madre, si trasferisce un campione su una matrice solida (membrana di nylon o nitrocellulosa) matrice Subcultura della cellule dalle colonie positive provenienti dalla piastra madre si lisano le cellule della matrice e si denatura il DNA liberato, deproteinizzandolo e legandolo irreversibilmente alla matrice Incubazione con la sonda di DNA complementare al DNA di interesse. Lavaggi che eliminano la sonda non legata. Autoradiografia che rivela i cloni identificati con la sonda
 matrice. Subcultura della cellule. dalle colonie positive. provenienti dalla piastra madre. si lisano le cellule della matrice e si denatura il DNA liberato, deproteinizzandolo e legandolo irreversibilmente alla matrice. Incubazione con la sonda di DNA. complementare al DNA di interesse. Lavaggi che eliminano la sonda non legata. Autoradiografia che rivela i cloni. identificati con la sonda.")
8
Screening di librerie geniche
2. Immunodosaggio di colonie 2. ciascuna colonia formatasi sulla piastra madre, si trasferisce un campione su una matrice solida (membrana di nylon o nitrocellulosa) 1. Trasferimento cellule matrice Piastra madre Subcultura della cellule dalla colonia positiva proveniente dalla piastra madre 3. si lisano le cellule della matrice e si ancorano le proteine alla matrice 4. Anticorpo primario (l) l 6. Eliminazione per lavaggio del secondario non legato e si effettua una reazione cromatica che funziona solo in presenza del secondario. l w 5. Eliminazione per lavaggio del primario e incubazione con anticorpo secondario (w) l w
 1. Trasferimento. cellule. matrice. Piastra. madre. Subcultura della cellule. dalla colonia positiva. proveniente dalla piastra madre. 3. si lisano le cellule della matrice e si ancorano le proteine alla matrice. 4. Anticorpo primario (l) l. 6. Eliminazione per lavaggio del secondario non legato e si effettua una reazione cromatica che funziona solo in presenza del secondario. l. w. 5. Eliminazione per lavaggio del primario e incubazione con anticorpo secondario (w) l. w.")
9
Esempio: HaeIII (da Haemophilus parainfluenzae)
Estremità piatte Esempio: HaeIII (da Haemophilus parainfluenzae) La sequenza di DNA in doppia elica: 5’-NNNNGGCCNNNN-3’ 3’-NNNNCCGGNNNN-5’ viene tagliata nel seguente modo: 5’-NNNNGG pCCNNNN-3’ 3’-NNNNCCp GGNNNN-5’ Estremità coesive Esempio: EcoRI La sequenza di DNA in doppia elica: 5’-NNNNGAATTCNNNN-3’ 3’-NNNNCTTAAGNNNN-5’ viene tagliata nel seguente modo: 5’-NNNNG pAATTCNNNN-3’ 3’-NNNNCTTAAp GNNNN-5’ In questo caso il taglio è perfettamente simmetrico. Le estremità piatte sono saldate in modo inefficiente dalla DNA ligasi, perché non restano associate a lungo. Tutte le estremità generate dagli enzimi che operano un taglio sfalsato, siano esse sporgenti in 5’ o in 3’, sono “appiccicose”, cioè possono formare ponti idrogeno tra le due code a filamento singolo complementari.
 La sequenza di DNA in doppia elica: 5’-NNNNGGCCNNNN-3’ 3’-NNNNCCGGNNNN-5’ viene tagliata nel seguente modo: 5’-NNNNGG pCCNNNN-3’ 3’-NNNNCCp GGNNNN-5’ Estremità coesive. Esempio: EcoRI. La sequenza di DNA in doppia elica: 5’-NNNNGAATTCNNNN-3’ 3’-NNNNCTTAAGNNNN-5’ viene tagliata nel seguente modo: 5’-NNNNG pAATTCNNNN-3’ 3’-NNNNCTTAAp GNNNN-5’ In questo caso il taglio è perfettamente simmetrico. Le estremità piatte sono saldate in modo inefficiente dalla DNA ligasi, perché non restano associate a lungo. Tutte le estremità generate dagli enzimi che operano un taglio sfalsato, siano esse sporgenti in 5’ o in 3’, sono appiccicose , cioè possono formare ponti idrogeno tra le due code a filamento singolo complementari.")
10
Rappresentazione schematica del clonaggio (II) - estremità coesive
annealing reannealing Tutti questi appaiamenti (annealing-reannealing) sono transitori a causa della debolezza dei legami a idrogeno tra il numero ridotto di basi a livello delle estremità coesive. Questi legami vengono stabilizzati grazie all’uso dell’enzima DNA LIGASI in un processo noto con il nome di ligazione. Enzima T4 DNA ligasi (isolato dal batteriofago T4) forma legami covalenti tra il gruppo fosfato 5’ di una estremità e il gruppo 3’ OH della catena adiacente. L’enzima lavora a 10°C e richiede ATP.
 sono transitori a causa della debolezza dei legami a idrogeno tra il numero ridotto di basi a livello delle estremità coesive. Questi legami vengono stabilizzati grazie all’uso dell’enzima DNA LIGASI in un processo noto con il nome di ligazione. Enzima T4 DNA ligasi (isolato dal batteriofago T4) forma legami covalenti tra il gruppo fosfato 5’ di una estremità e il gruppo 3’ OH della catena adiacente. L’enzima lavora a 10°C e richiede ATP.")
11
Rappresentazione schematica del clonaggio (III) - estremità coesive
 - estremità coesive")
12
Trattamento del DNA con estremità piatte
La ligazione di frammenti con estremità piatte non è efficiente quanto quella di frammenti con estremità coesive, perciò prima di unire il DNA con il vettore di clonaggio è necessario eseguire alcune procedure aggiuntive: AGGIUNTA DEL LINKER plasmide DNA a estremità piatte 5’ pGGGATCCC 3 CCCTAGGG linker Unione del linker al DNA con estramità piatte mediante DNA ligasi 5’ pGGGATCCC 3’ CCCTAGGG GGGATCCC CCCTAGGG Taglio con BamHI CTAGp 5’pGATC 3’ 5’ pGATCCC 3’ GG GG CCCTAGp Taglio con BamHI IN ALTRI CASI L POTO DEL LINKER SI PUò AGGIUNGERE UN ADATTAORE CHE HA GIA ESTREMITà COESIVE COSI DA NON PASSARE PER LA DIGESTIONE ENZIMATICA C T A G G G G G G A T C G A T C C C C C C T A G Unione delle estremità coesive mediante DNA ligasi

13
Vettori per il clonaggio (I)
Per clonare una qualsiasi molecola di DNA è necessario inserirla in un vettore per il clonaggio. Questi elementi di DNA possono essere mantenuti e propagati in un organismo ospite in cui il vettore possa replicarsi. Un tipico ospite è Escherichia Coli che cresce e si divide rapidamente. Qualsiasi vettore con un origine di replicazione adatta si replica con successo in E.Coli (assieme al DNA inserito). Quindi, qualsiasi DNA inserito in un vettore può essere amplificato (se ne possono produrre quantità illimitate) e usato per successive analisi Libreria genica: ogni vettore contiene un frammento diverso di DNA estraneo Isolamento di un clone mediante screening della libreria Dire che I vettori che si utilizzano sono diversi e differiscon per complessità, selezione e lunghezza di DNA che si può inserire. In genere I vettori sono stati sviluppati da molecole presenti in natura Amplificazione del singolo clone
. Quindi, qualsiasi DNA inserito in un vettore può essere amplificato (se ne possono produrre quantità illimitate) e usato per successive analisi. Libreria genica: ogni vettore contiene un frammento diverso di DNA estraneo. Isolamento di un clone mediante screening della libreria. Dire che I vettori che si utilizzano sono diversi e differiscon per complessità, selezione e lunghezza di DNA che si può inserire. In genere I vettori sono stati sviluppati da molecole presenti in natura. Amplificazione. del singolo clone.")
14
Vettori per il clonaggio (II)
- SONO STATI SVILUPPATI DA MOLECOLE PRESENTI IN NATURA (PLASMIDI BATTERICI, BATTERIOFAGI) O COMBINAZIONE DEGLI ELEMENTI CHE LI COMPONGONO (COSMIDI). Plasmidi (i) DNA extracromosomico circolare di piccole dimensioni presente in molti batteri che conferisce resistenza a antibiotici, capacità di metabolizzare substrati e capacità di coniugazione. anni ’70: alcuni di questi vettori sono stati modificati in laboratorio (digestioni e successive ligazioni) pr costruire vettori di clonaggio (es: pBR322 da Bolivar e Rodriguez); un plasmide artificiale è molto più piccolo di quello naturale e viene più facilmente inserito nei batteri nel processo noto come trasformazione; un origine di replicazione del DNA di tipo batterico cosicchè il DNA può essere replicato dalla cellula ospite plasmidi come pBR322 hanno un origine di replicazione rilassata, che non segue cioè la divisione cellulare, in modo che la replicazione del plasmide è molto più frequente di quella cromosomica. In questo modo ogni cellula produce molte molecole di plasmide. Sono stati introdotti due geni che codificano per la rsistenza agli antibiotici in vari punti del palsmide ci sono siti singoli di riconoscimento per una serie di enzimi di restrizione. Alcuni di questi siti possono essere inseriti in un gene che codifica per la resistenza ad un antibiotico, cosicchè la presenza di un particolare inserto può essere rivelata dalla perdita di resistenza a quello stesso antibiotico (inattivazione inserzionale).
 O COMBINAZIONE DEGLI ELEMENTI CHE LI COMPONGONO (COSMIDI). Plasmidi (i) DNA extracromosomico circolare di piccole dimensioni presente in molti batteri che conferisce resistenza a antibiotici, capacità di metabolizzare substrati e capacità di coniugazione. anni ’70: alcuni di questi vettori sono stati modificati in laboratorio (digestioni e successive ligazioni) pr costruire vettori di clonaggio (es: pBR322 da Bolivar e Rodriguez); un plasmide artificiale è molto più piccolo di quello naturale e viene più facilmente inserito nei batteri nel processo noto come trasformazione; un origine di replicazione del DNA di tipo batterico cosicchè il DNA può essere replicato dalla cellula ospite. plasmidi come pBR322 hanno un origine di replicazione rilassata, che non segue cioè la divisione cellulare, in modo che la replicazione del plasmide è molto più frequente di quella cromosomica. In questo modo ogni cellula produce molte molecole di plasmide. Sono stati introdotti due geni che codificano per la rsistenza agli antibiotici. in vari punti del palsmide ci sono siti singoli di riconoscimento per una serie di enzimi di restrizione. Alcuni di questi siti possono essere inseriti in un gene che codifica per la resistenza ad un antibiotico, cosicchè la presenza di un particolare inserto può essere rivelata dalla perdita di resistenza a quello stesso antibiotico (inattivazione inserzionale).")
15
Vettori per il clonaggio (III)
pBR322 Esempio di mutagenesi inserzionale (singolo taglio con BamHI)
")
16
Inattivazione Inserzionale
pBR322 dig BamHI singolo sito di restrizione Frammento di DNA cromosomico dig BamHI + Plasmide contenente l’inserto Plasmide ricircolarizzato Plasmide che contiene più copie del frammento T4 DNA LIGASI TRASFORMAZIONE: incorporazione di piccole molecole circolari in E.Coli colonie cresciute su piastra contenente Ampicillina, crescono solo le cellule che contengono il plasmide Recupero delle colonie contenenti plasmide+inserto presenti sulla piastra AmpR Piastramento in replica utilizzando un tampone di velluto sterile Piastra contenente Tetraciclina Non è possibile allo stadio Amp distinguere le colonie che posseggono I l plasmide richiuso o il plasmide con l’inserto. Si fa un piastramento in replica utilizzando un tampone di velluto sterile su piastre contenente terreno con tetraciclina crescono solo le cellule con plasmide senza insertto: incubazione

17
Plasmidi (ii) : pUC gene per la resistenza alla tetraciclina;
origine di replicazione per E.Coli; Multiple Cloning Site (MCS) o polylinker: siti di restrizione unici più comuni; MCS è parte del gene lacZ che codifica per la b-galattosidasi;
 o polylinker: siti di restrizione unici più comuni; MCS è parte del gene lacZ che codifica per la b-galattosidasi;")
18
Plasmidi (iii) : pUC MCS Vettori non ricombinanti (senza inserto)
Quando si trasformano cellule di E.Coli con il plasmide pUC, il gene può essere attivato aggiungnedo nel terreno l’induttore isopropil-b-D-tiogalattopiranoside (IPTG), la cui presenza induce l’espressione della b-galattosidasi. L’ezima funzionale idrolizza una sostanza incolore detta 5-bromo-4-cloro-3-indolil-b-galattopiranoside (X-Gal) producendo un composto blu insolubile. Vettori non ricombinanti (senza inserto) MCS GENE PER LA b-GALATTOSIDASI X-Gal idrolizzato: placca blu INDUZIONE CON IPTG 2. Vettori ricombinanti (senza inserto) INSERIMENTO DEL DNA NEL MCS CON INTERRUZIONE DEL GENE b-GAL INDUZIONE CON IPTG GENE INSERITO X-Gal NON idrolizzato: placca bianca
, la cui presenza induce l’espressione della b-galattosidasi. L’ezima funzionale idrolizza una sostanza incolore detta 5-bromo-4-cloro-3-indolil-b-galattopiranoside (X-Gal) producendo un composto blu insolubile. Vettori non ricombinanti (senza inserto) MCS. GENE PER LA b-GALATTOSIDASI. X-Gal idrolizzato: placca blu. INDUZIONE CON IPTG. 2. Vettori ricombinanti (senza inserto) INSERIMENTO DEL DNA NEL MCS CON INTERRUZIONE DEL GENE b-GAL. INDUZIONE CON IPTG. GENE INSERITO. X-Gal NON idrolizzato: placca bianca.")
19
Vettori derivati da virus (I)
Per inserti lunghi (più di 5kb) si usano vettori derivanti dal batteriofago l in quanto offrono una efficienza di clonaggio superiore di circa 16 volte rispetto ai vettori plasmidici; il batteriofago l è un fago con DNA lieare a doppia elica, lungo circa 49 kb in grado di infettare E.Coli, inserendogli il proprio DNA attraverso la membrana cellulare il DNA del fago l può replicarsi secondo due modalità: si integra nel cromosoma di E. Coli dove resta quiesciente fino a quando un segnale ne promuove l’escissione (ciclo lisogenico) si replica aapena infetta la cellula, sintetizza le proteine della testa e della coda, si formano nuove particelle fagiche (ciclo litico) PER GLI INSERTI LUNGHI (GIA QUELLI DI 5KB) AUMENATO TANTO LE DIMENSIONI DEI PLASMIDI AL PUNTO CHE L’EFFICIENZA DELLA TRASFORMAZIONE DELLE CELLULE BATTERIUCHE DIMINUISCE MOLTO
 si usano vettori derivanti dal batteriofago l in quanto offrono una efficienza di clonaggio superiore di circa 16 volte rispetto ai vettori plasmidici; il batteriofago l è un fago con DNA lieare a doppia elica, lungo circa 49 kb in grado di infettare E.Coli, inserendogli il proprio DNA attraverso la membrana cellulare. il DNA del fago l può replicarsi secondo due modalità: si integra nel cromosoma di E. Coli dove resta quiesciente fino a quando un segnale ne promuove l’escissione (ciclo lisogenico) si replica aapena infetta la cellula, sintetizza le proteine della testa e della coda, si formano nuove particelle fagiche (ciclo litico) PER GLI INSERTI LUNGHI (GIA QUELLI DI 5KB) AUMENATO TANTO LE DIMENSIONI DEI PLASMIDI AL PUNTO CHE L’EFFICIENZA DELLA TRASFORMAZIONE DELLE CELLULE BATTERIUCHE DIMINUISCE MOLTO.")
20
Vettori derivati da virus (II)
coda DNA testa I fagi sono stati utilizzati per la costruzione di librerie geniche perchè hanno permesso di introdurre frammenti più grandi di DNA rispetto ai plasmidi. Il DNA del batteriofago l è lungo circa 50 kb, approssimativamente 20 dei quali risultano indispensabili agli eventi di integrazione-escissione (I/E). Per formare le genoteche si è ragionato sulla possibilità di sostituire queste 20 kb con altrettante kb di DNA clonato. Una simile molecola di DNA si potrebbe perpetuare sotto forma di batteriofago l “ricombinante” tramite cicli litici forzosi.
. Per formare le genoteche si è ragionato sulla possibilità di sostituire queste 20 kb con altrettante kb di DNA clonato. Una simile molecola di DNA si potrebbe perpetuare sotto forma di batteriofago l ricombinante tramite cicli litici forzosi.")
21
Cosmidi Vettori di clonaggio Vector system Host cell
Possono trasportare 40 kb di DNA clonato e possono essere mantenuti come plasmidi in E. Coli. Vettori di clonaggio Vector system Host cell Insert capacity (kb) Plasmid E. coli 0.1-10 Bacteriophage l 10-20 Cosmid 35-45 Bacteriophage P1 80-100 BAC (bacterial artificial chromosome) 50-300 P1 bacteriophage-derived AC YAC Yeast 100-2,000
 Plasmid. E. coli Bacteriophage l Cosmid Bacteriophage P BAC (bacterial artificial chromosome) P1 bacteriophage-derived AC YAC. Yeast ,000.")
22
La trasformazione (I) introduzione del DNA nella
cellula ospite (trasformazione) e isolamento delle cellule con il gene clonato Produzione della proteina dal gene clonato proteina codificata dal gene clonato
 e isolamento delle cellule con il gene clonato. Produzione della proteina. dal gene clonato. proteina codificata. dal gene clonato.")
23
La trasformazione (II)
TRASFORMAZIONE: assunzione di DNA da parte di un organismo 1970: Mandel and Higa osservarono che batteri, trattati con soluzioni di CaCl2 e riscaldati velocemente, potevano essere trasformati con DNA del batteriofago λ quindi questo trattamento conferiva ai batteri un transiente stato di “competenza” COMPETENZA: capacità di assumere DNA = numero di colonie positive/microgrammo di DNA trasformato Nel corso degli anni sono stati fatti diversi tentativi per migliorare l’efficienza di trasformazione. Le due tecniche maggiormente utilizzate oggi sono: Trasformazione per CaCl2 Elettroporazione

24
Trasformazione Per Elettroporazione
Le cellule batteriche unite al DNA vengono sottoposte ad una veloce scarica elettrica che permette la formazione di “elettropori” che favoriscono l’entrata di DNA. Competenza: cellule trasformate /μg di DNA

25
Trasformazione in CaCl2
Crescita di un ceppo E.Coli in terreno ricco di nutrienti. Le cellule si raccolgono quando sono in fase logaritmica

26
I cationi divalenti schermano le cariche negative sul DNA (dei gruppi fosfato)
La combinazione di bassa temperatura e di cationi divalenti può aiutare a cristallizzare regioni della membrana rendendo più accessibili i canali di assunzione del DNA

27
Sommario delle fasi importanti nella clonazione del DNA
Vettore plasmidico e DNA esogeno da inserire devono avere estremità compatibili. I due “pezzi” di DNA sono fusi creando molecole di DNA ricombinante; Introduzione del DNA in cellule batteriche ospiti idonee (trasformazione) Selezione delle cellule resistenti all’antibiotico e selezione dei cloni che hanno assunto il DNA ricombinante
 Selezione delle cellule resistenti all’antibiotico e selezione dei cloni che hanno assunto il DNA ricombinante.")
28
Primo esempio di clonaggio
gene di interesse BamHI EcoRI VETTORE I

29
Secondo esempio di clonaggio
gene di interesse Not1 BglII VETTORE I …Come si procede ???... se decido di clonare il gene di interesse BamHI-EcoRI devo essere sicura che gli stessi enzimi non tagliano all’interno della mia sequenza genica; Devo disegnare due oligo, uno frw con la sequenza che possiede il sito di riconoscimento per l’enzima BamHI e uno reverse che contiene il sito di riconoscimento per l’enzima EcoRI Amplificazione per PCR della sequenza genica di interesse con gli oligo disegnati; Purificazione della PCR preparativa Digestione con gli enzimi BamHI e EcoRI per la formazione delle estremità coesive Digestione del vettore pOTB7 con BamH1 e EcoRI Ligazione con l’ezima T4 DNA ligasi

30
1 atggagatgg aaaaggagtt cgagcagatc gacaagtccg ggagctgggc ggccatttac
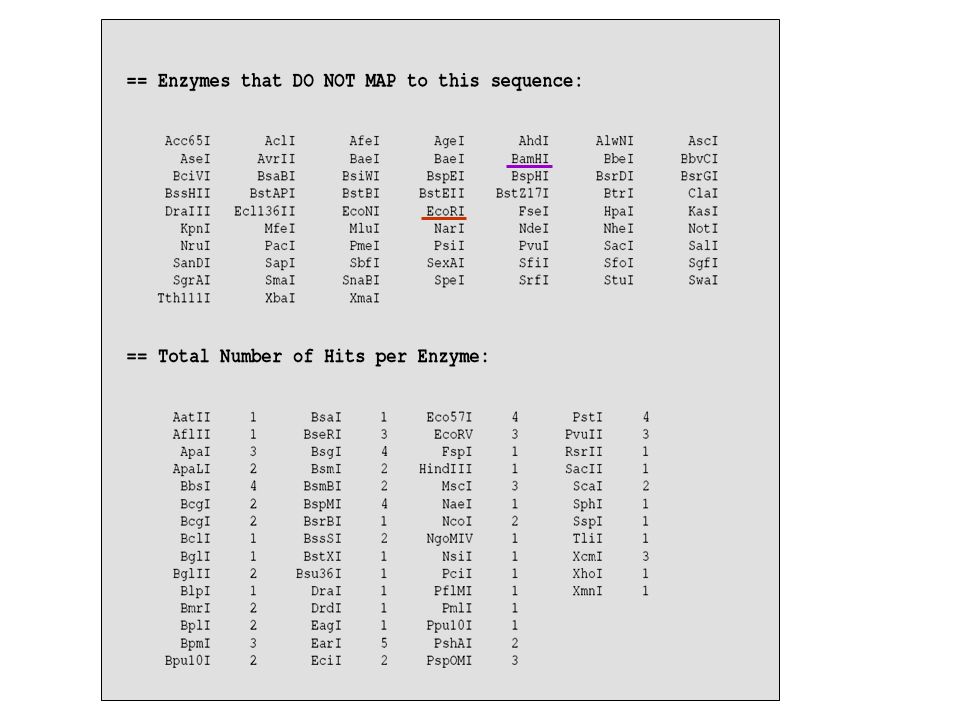
se decido di clonare il gene di interesse BamHI-EcoRI devo essere sicura che gli stessi enzimi non tagliano all’interno della mia sequenza genica; 1 atggagatgg aaaaggagtt cgagcagatc gacaagtccg ggagctgggc ggccatttac 61 caggatatcc gacatgaagc cagtgacttc ccatgtagag tggccaagct tcctaagaac 121 aaaaaccgaa ataggtacag agacgtcagt ccctttgacc atagtcggat taaactacat 181 caagaagata atgactatat caacgctagt ttgataaaaa tggaagaagc ccaaaggagt 241 tacattctta cccagggccc tttgcctaac acatgcggtc acttttggga gatggtgtgg 301 gagcagaaaa gcaggggtgt cgtcatgctc aacagagtga tggagaaagg ttcgttaaaa 361 tgcgcacaat actggccaca aaaagaagaa aaagagatga tctttgaaga cacaaatttg 421 aaattaacat tgatctctga agatatcaag tcatattata cagtgcgaca gctagaattg 481 gaaaacctta caacccaaga aactcgagag atcttacatt tccactatac cacatggcct 541 gactttggag tccctgaatc accagcctca ttcttgaact ttcttttcaa agtccgagag 601 tcagggtcac tcagcccgga gcacgggccc gttgtggtgc actgcagtgc aggcatcggc 661 aggtctggaa ccttctgtct ggctgatacc tgcctcttgc tgatggacaa gaggaaagac 721 ccttcttccg ttgatatcaa gaaagtgctg ttagaaatga ggaagtttcg gatggggctg 781 atccagacag ccgaccagct gcgcttctcc tacctggctg tgatcgaagg tgccaaattc 841 atcatggggg actcttccgt gcaggatcag tggaaggagc tttcccacga ggacctggag 901 cccccacccg agcatatccc cccacctccc cggccaccca aacgaatcct ggagccacac 961 aatgggaaat gcagggagtt cttcccaaat caccagtggg tgaaggaaga gacccaggag 1021 gataaagact gccccatcaa ggaagaaaaa ggaagcccct taaatgccgc accctacggc 1081 atcgaaagca tgagtcaaga cactgaagtt agaagtcggg tcgtgggggg aagtcttcga 1141 ggtgcccagg ctgcctcccc agccaaaggg gagccgtcac tgcccgagaa ggacgaggac 1201 catgcactga gttactggaa gcccttcctg gtcaacatgt gcgtggctac ggtcctcacg 1261 gccggcgctt acctctgcta caggttcctg ttcaacagca acacatag Programma che permette di fare un’analisi di restrizione della sequenza

32
CTAGCGGATCCGGCCCGTCATGGAGATGGAA
2. Devo disegnare due oligo, uno frw con la sequenza che possiede il sito di riconoscimento per l’enzima BamHI e uno reverse che contiene il sito di riconoscimento per l’enzima EcoRI ggcccgtcatggagatggaaaaggagttcgagcagatcgacaagtccgggagctgggcggccatttac gccggcgcttacctctgctacaggttcctgttcaacagcaacacatag Costruzione dell’oligo frw Codina di 5-6 nt - sequenza riconosciuta dall’enzima di restrizione (BamHI) – sequenza compresa di atg (codone di inizio). L’oligo deve essere lungo circa nt CTAGCGGATCCGGCCCGTCATGGAGATGGAA Costruzione dell’oligo reverse Sequenza fino al codone di stop (stop incluso)- sequenza riconosciuta dall’enzima di restrizione- e codina di 5-6 nt. Fare il reverse complement TTCCTGTTCAACAGCAACACATAGGAATTCCTAGC GCTAGGAATTCCTATGTGTTGCTGTTGAACAGGAA
 – sequenza compresa di atg (codone di inizio). L’oligo deve essere lungo circa nt. CTAGCGGATCCGGCCCGTCATGGAGATGGAA. Costruzione dell’oligo reverse. Sequenza fino al codone di stop (stop incluso)- sequenza riconosciuta dall’enzima di restrizione- e codina di 5-6 nt. Fare il reverse complement. TTCCTGTTCAACAGCAACACATAGGAATTCCTAGC. GCTAGGAATTCCTATGTGTTGCTGTTGAACAGGAA.")
33
Purificazione della PCR preparativa
CTAGCGGATCCGGCCCGTCATGGAGATGGAA CCGGGCAGTACCTCTACCTTTTCCTC GGCCCGTCATGGAGATGGAAAAGGAGTTCGAGCAGATCGACAAGTCCGGGAGCTGGGCGGCCATTTAC CGGCCGCGAATGGAGACGATGTCCAAGGACAAGTTGTCGTTGTGTATC GCCGGCGCTTACCTCTGCTACAGGTTCCTGTTCAACAGCAACACATAG AAGGACAAGTTTGTCGTTGTGTATCCTTAAGGATCG Amplificazione per PCR della sequenza genica di interesse con gli oligo disegnati; Purificazione della PCR preparativa Digestione con gli enzimi BamHI e EcoRI per la formazione delle estremità coesive Digestione del vettore pOTB7 con BamH1 e EcoRI Ligazione con l’ezima T4 DNA ligasi

Presentazioni simili