Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Di che cosa trattiamo? Alcuni cenni sulla struttura delle PROTEINE
L’analisi informatica per la comprensione della struttura e funzione delle proteine: le SIMULAZIONI AL CALCOLATORE

2
La struttura delle biomolecole è un argomento di grande interesse per molti settori della ricerca di base e applicativa Funzione STRUTTURA Meccanismo Origine/Evoluzione

3
La FISICA fornisce metodi computazionali e approcci sperimentali molto potenti per indagare la struttura delle macromolecole biologiche.

4
Le più importanti tecniche sperimentali e teoriche per l’analisi delle strutture di macromolecole
Difrattometria ai raggi X Microscopie Spettroscopie Modellistica molecolare Simulazioni al computer di dinamiche molecolare e interazioni molecolari

5
Le proteine e gli acidi nucleici sono polimeri
Le macromolecole sono costruite collegando insieme unità strutturali definite MONOMERI Proteine 20 amminoacidi 4 basi nucleotidiche Acidi nucleici

6
La luce prodotta dalle lucciole coinvolge la proteina luciferina;
LE PROTEINE (dal greco protos: principale) SONO LE MACROMOLECOLE PIU’ ABBONDANTI E SVOLGONO SVARIATE FUNZIONI, ad es.: La luce prodotta dalle lucciole coinvolge la proteina luciferina; Gli eritrociti contengono la proteina emoglobina che trasporta l’ossigeno; La proteina cheratina è il componente strutturale dei capelli, squame, corna, lana, unghie etc.
 SONO LE MACROMOLECOLE PIU’ ABBONDANTI E SVOLGONO SVARIATE FUNZIONI, ad es.: La luce prodotta dalle lucciole coinvolge la proteina luciferina; Gli eritrociti contengono la proteina emoglobina che trasporta l’ossigeno; La proteina cheratina è il componente strutturale dei capelli, squame, corna, lana, unghie etc.")
7
Contengono 20 amminoacidi uniti tramite un legame “peptidico”covalente
Amminoacido Il legame peptidico

8
La sequenza degli amminoacidi costituisce la struttura primaria
Ogni pallina corrisponde ad un amminoacido Ribonucleasi: Mr da, 124 aa., secreta dal pancreas. Catalizza l’idrolisi di acidi nucleici ingeriti con la dieta

9
Le proteine hanno diversi livelli di struttura

10
Le principali strutture secondarie di una catena polipetidica: elica…
MODELLO A PALLE E BASTONCINI Elica destrorsa, Si evidenziano i legami idrogeno Il passo dell’elica è 5.4 Å o 3.6 residui amminoacidici

11
…e foglietto I legami idrogeno si formano tra segmenti adiacenti
I segmenti adiacenti possono anche essere lontani nella sequenza amminoacidica Le catene possono essere parallele o antiparallele

12
Classificazione generale delle strutture terziarie
Proteine con predominanza di elica Proteine miste Proteine con predominanza di sheets

13
La struttura quaternaria delle proteine
La struttura quaternaria riguarda proteine costituite da più catene polipeptidiche o da più domini strutturali (es. proteine regolatrici) Le interazioni tra le subunità consentono grandi variazioni nell’attività catalitica Esempio: la emoglobina
 Le interazioni tra le subunità consentono grandi variazioni nell’attività catalitica. Esempio: la emoglobina.")
14
Le interazioni non covalenti che partecipano nella definizione della struttura delle biomolecole

15
Molte malattie sono dovute al difettoso ripiegamento di una proteina
Alcune patologie derivano da proteine che non sono in grado di raggiungere la loro struttura funzionale e che tendono a formare grossi aggregati (fibrille o forme amiloidi): Alzheimer, Parkinson, encefalopatia spongiforme, diabete di tipo II. In altri casi mutazioni puntiformi generano proteine che non raggiungono la loro locazione finale o che non sono più in grado di svolgere la loro funzione perché incapaci di legare i loro substrati. Fibrosi cistica: difetto nella proteina transmembrana che agisce come un canale degli ioni cloro nelle cellule epiteliali (CFTR: 1480 amminoacidi). La mutazione più comune è la delezione di un amminoacido (Phe 508) e la proteina mutata non si avvolge correttamente.
: Alzheimer, Parkinson, encefalopatia spongiforme, diabete di tipo II. In altri casi mutazioni puntiformi generano proteine che non raggiungono la loro locazione finale o che non sono più in grado di svolgere la loro funzione perché incapaci di legare i loro substrati. Fibrosi cistica: difetto nella proteina transmembrana che agisce come un canale degli ioni cloro nelle cellule epiteliali (CFTR: 1480 amminoacidi). La mutazione più comune è la delezione di un amminoacido (Phe 508) e la proteina mutata non si avvolge correttamente.")
16
Il problema del “folding”: che cosa determina la struttura tridimensionale delle proteine?
In genere la sequenza amminoacidica contiene la informazione completa per il raggiungimento della struttura funzionale

17
DETERMINAZIONE DELLA STRUTTURA TRIDIMENSIONALE DELLE MACROMOLECOLE BIOLOGICHE A LIVELLO ATOMICO
= determinare le coordinate spaziali xyz per ogni singolo atomo, ovvero le posizioni reciproche degli atomi nello spazio

18
TECNICHE SPERIMENTALI
X-ray crystallography (Kendrew and Perutz ) Diffrazione di raggi X incidenti su cristalli di biomolecole -> dipende dalla spaziatura tra gli atomi NMR spectroscopy (Ernst and Wuthrich ) Assorbimento di radio frequenze da parte di biomolecole (solo certi tipi di nuclei) IN SOLUZIONE e immerse in un campo magnetico -> dipende dall’intorno del nucleo
 Diffrazione di raggi X incidenti su cristalli di biomolecole -> dipende dalla spaziatura tra gli atomi. NMR spectroscopy. (Ernst and Wuthrich ) Assorbimento di radio frequenze da parte di biomolecole (solo certi tipi di nuclei) IN SOLUZIONE e immerse in un campo magnetico -> dipende dall’intorno del nucleo.")
19
Confronto NMR - diffrazione ai raggi X
Mioglobina Con NMR si ottengono delle famiglie di strutture: si hanno informazioni sulla mobilità delle porzioni di macromolecola, ma in molti casi il risultato finale è molto simile

20
TECNICHE COMPUTAZIONALI
L’utilizzo COMPLEMENTARE di tecniche di tipo sperimentale e di tipo computazionale è l’approccio ottimale per lo studio dei sistemi e dei processi biologici. I limiti dell’una sono infatti superati dall’altra

21
Limiti delle tecniche sperimentali
risoluzione SPAZIALE misure ad alta risoluzione di strutture molecolari sono possibili solo per sistemi relativamente rigidi risoluzione ENERGETICA analisi delle energie di interazione atomica difficoltosa risoluzione TEMPORALE i primissimi eventi dei processi biologici sono di difficile misurazione

22
Limiti delle tecniche computazionali
Sistemi biomolecolari troppo complessi si devono utilizzare approssimazioni Simulazione del comportamento nel tempo di un sistema molecolare su un computer solo un numero limitato (<NA) atomi o di gradi di libertà (di solito atomi), per un limitato periodo di tempo ( picosecondi) può essere simulato piccoli sistemi, con tempi di rilassamento brevi e/o utilizzo dei dati sperimentali per restringere il numero di conformazioni possibili
 atomi o di gradi di libertà (di solito atomi), per un limitato periodo di tempo ( picosecondi) può essere simulato piccoli sistemi, con tempi di rilassamento brevi e/o utilizzo dei dati sperimentali per restringere il numero di conformazioni possibili.")
23
Scala dei tempi e delle distanze per una proteina
Å s Movimenti locali: fluttuazioni atomiche movimenti delle catene laterali movimenti dei loops Å s Movimenti di corpo rigido: movimento delle eliche movimento dei domini (piegamento degli hinge loops) movimento delle subunità > 5 Å s Movimenti su larga scala: transizioni elica - coil dissociazione/associazione folding e unfolding
 movimento delle subunità. > 5 Å s Movimenti su larga scala: transizioni elica - coil. dissociazione/associazione. folding e unfolding.")
24
Alcune Applicazioni MODELLIZZARE la struttura tridimensionale a risoluzione atomica della molecola

25
Effettuare MUTAZIONI puntiformi, che possono fornire indicazioni utili per il riconoscimento del sito attivo o di strutture indispensabili all'attività della molecola o dirette ad una certa funzione.

26
Studiare le VARIAZIONI CONFORMAZIONALI provocate dall’interazione della proteina con uno o più LIGANDI, la quale fornisce l’attivazione (o inattivazione) necessaria per compiere la propria funzione biologica (o per impedirla).
 necessaria per compiere la propria funzione biologica (o per impedirla).")
27
Monitorare i CAMBIAMENTI STRUTTURALI indotti su peptidi o proteine da parte di MEMBRANE BIOLOGICHE, i quali sembrano essere fondamentali per il riconoscimento con il recettore o per oltrepassare la fase lipidica e raggiungere zone altrimenti inaccessibili.

28
Comprendere il processo di FOLDING delle proteine, ovvero il meccanismo di ripiegamento con cui raggiungono la confomazione biologicamente attiva.

29
Applicazioni FARMACOLOGICHE: viene fornita un’indicazione specifica, o quanto meno restrittiva, della struttura opportuna in funzione del bersaglio del farmaco. In questo campo, la costruzione di strutture calibrate permette di ridurre la ricerca ad un ristretto raggio d’azione.

30
Modellizzazione Molecolare

31
Ipotesi termodinamica di Anfinsen (per proteine a singolo dominio)
L’informazione codificata nella sequenza amminoacidica di una proteina determina completamente la sua struttura nativa Lo stato nativo è il minimo assoluto dell’energia libera della proteina

32
Sequenze casuali non sono in grado di ripiegarsi in maniera univoca
Le proteine presenti in natura non sono sequenze casuali. Esse sono una piccola famiglia di sequenze selezionate dalla natura attraverso l’evoluzione. Ciascuna ha un minimo globale distinto, ben separato dagli altri stati metastabili. Quali sono le proprietà uniche e comuni a tutti gli elementi di questo particolare insieme di sequenze di tipo proteico?

33
Diagramma di flusso della modellizzazione proteica
Sequenza proteica Dati sperimentali Ricerca nelle banchedati Allineamento multiplo di sequenza Assegnazione dei domini Proteina omologa nella banca dati PDB? Predizione della struttura secondaria Predizione del fold No E’ stato predetto un fold? Analisi della famiglia del fold Sì Sì Allineamento delle strutture secondarie Allineamento della sequenza alla struttura Modellizzazione comparativa No Predizione della struttura terziaria Modello tridimensionale della proteina

34
Modellizzazione comparativa (o per similarità di sequenza)
Permette di costruire la struttura tridimensionale di una proteina sulla base della SIMILARITÀ DI SEQUENZA con un’altra proteina di struttura NOTA che viene usata come STAMPO.

35
Si basa sulle seguenti osservazioni:
le proteine appartengono ad un numero limitato di famiglie strutturali proteine della stessa famiglia hanno strutture tridimensionali molto simili

36
E’ molto importante l’allineamento di sequenza con la proteina “stampo”
* aa identici . aa simili

37
Dopodichè si procede con…
…la costruzione dello scheletro… …l’inserimento delle catene laterali… …l’inserimento dei loop corrispondenti a “buchi” nell’allineamento…

38
…per concludere con… …l’ottimizzazione del modello..
…e il controllo della sua qualità

39
Esempio: Deossiemoglobina umana (1a3n)
Il file PDB Esempio: Deossiemoglobina umana (1a3n) HEADER OXYGEN TRANSPORT JAN A3N TITLE DEOXY HUMAN HEMOGLOBIN COMPND MOL_ID: 1; COMPND 2 MOLECULE: HEMOGLOBIN; COMPND 3 CHAIN: A, B, C, D; COMPND 4 BIOLOGICAL_UNIT: ALPHA-BETA-ALPHA-BETA TETRAMER SOURCE MOL_ID: 1; SOURCE 2 ORGANISM_SCIENTIFIC: HOMO SAPIENS; SOURCE 3 ORGANISM_COMMON: HUMAN; SOURCE 4 TISSUE: BLOOD; SOURCE 5 CELL: RED CELL KEYWDS OXYGEN TRANSPORT, HEME, RESPIRATORY PROTEIN, ERYTHROCYTE EXPDTA X-RAY DIFFRACTION AUTHOR J.TAME,B.VALLONE REVDAT APR-98 1A3N REMARK REMARK REMARK 2 RESOLUTION ANGSTROMS REMARK 3 […]
 HEADER OXYGEN TRANSPORT 22-JAN-98 1A3N TITLE DEOXY HUMAN HEMOGLOBIN COMPND MOL_ID: 1; COMPND 2 MOLECULE: HEMOGLOBIN; COMPND 3 CHAIN: A, B, C, D; COMPND 4 BIOLOGICAL_UNIT: ALPHA-BETA-ALPHA-BETA TETRAMER SOURCE MOL_ID: 1; SOURCE 2 ORGANISM_SCIENTIFIC: HOMO SAPIENS; SOURCE 3 ORGANISM_COMMON: HUMAN; SOURCE 4 TISSUE: BLOOD; SOURCE 5 CELL: RED CELL KEYWDS OXYGEN TRANSPORT, HEME, RESPIRATORY PROTEIN, ERYTHROCYTE EXPDTA X-RAY DIFFRACTION AUTHOR J.TAME,B.VALLONE REVDAT 1 29-APR-98 1A3N 0 REMARK 1 REMARK 2 REMARK 2 RESOLUTION. 1.8 ANGSTROMS. REMARK 3. […]")
40
coordinate tipo di atomo tipo di amminoacido X Y Z …
ATOM N VAL A N ATOM CA VAL A C ATOM C VAL A C ATOM O VAL A O ATOM CB VAL A C ATOM CG1 VAL A C ATOM CG2 VAL A C ATOM N LEU A N ATOM CA LEU A C ATOM C LEU A C ATOM O LEU A O ATOM CB LEU A C ATOM CG LEU A C ATOM CD1 LEU A C ATOM CD2 LEU A C ATOM N SER A N ATOM CA SER A C ATOM C SER A C ATOM O SER A O ATOM CB SER A C ATOM OG SER A O …

41
Deossiemoglobina catena A
1a3n Deossiemoglobina catena A C O N S EME Fe

42
RASMOL v 2.7

43
Analisi computazionale
Comprende tecniche come: la DINAMICA MOLECOLARE, la RICERCA DEL MINIMO DELL’ENERGIA CONFORMAZIONALE, e il RICONOSCIMENTO MOLECOLARE (DOCKING).
.")
44
Assunzioni fondamentali
Nucleo ed elettroni sono «ammassati» in una sola particella atomo-simile. Tali particelle atomo-simili sono sferiche e possiedono una carica. Le interazioni sono basate sui potenziali delle molle e sui potenziali classici. Le interazioni sono parametrizzate per specifici set di atomi. Le interazioni determinano la DISTRIBUZIONE SPAZIALE delle particelle atomo-simili e la loro ENERGIA.

45
Ad ogni conformazione molecolare è associata una
ENERGIA POTENZIALE

46
La forma più semplice DELL’ENERGIA POTENZIALE di una molecola è :
Energia di ALLUNGAMENTO dei legami + Energia di PIEGAMENTO degli angoli di legame + Energia di TORSIONE degli angoli diedri + Energia delle interazioni di NON-LEGAME: repulsioni steriche, interazioni di Van der Waals, interazioni elettrostatiche bending stretching torsion ENERGIA pot =

47
Forma matematica più comune:
|-Energia di allungamento-| | Energia di piegamento | | Energia torsionale | | Energia di non-legame |

48
Minimizzazione dell'energia
Minimizzare l'energia potenziale di una molecola significa trovare un percorso (costituito dalle variazioni dei gradi di libertà intramolecolari) che conduca da una conformazione iniziale alla conformazione a minima energia più vicina (MINIMO LOCALE), usando il minor numero di calcoli possibile.
 che conduca da una conformazione iniziale alla conformazione a minima energia più vicina (MINIMO LOCALE), usando il minor numero di calcoli possibile.")
49
Ipotesi termodinamica di Anfinsen (per proteine a singolo dominio)
L’informazione codificata nella sequenza amminoacidica di una proteina determina completamente la sua struttura nativa Lo stato nativo è il minimo assoluto dell’energia libera della proteina

50
Termodinamica del folding visto come imbuto di energia libera
Si raggiunge il minimo energetico assoluto percorrendo anche strade diverse Ogni minimo relativo corrisponde a stati intermedi metastabili Le interazioni idrofobiche di residui non polari provocano il “collasso idrofobico” Scendendo verso il basso si ha una diminuzione delle specie

51
Strategie di ricerca del minimo assoluto
Esistono metodi computazionali di ricerca del minimo assoluto che implicano il campionamento dello spazio conformazionale: il campionamento energetico sistematico l’annealing simulato la ricerca casuale enorme onere computazionale utilizzabili solo per peptidi corti

52
Dinamica molecolare Permette lo studio di processi dinamici complessi che avvengono nei sistemi biologici. Studia sia transizioni conformazionali che vibrazioni locali, ad esempio: stabilità delle proteine variazioni conformazionali folding proteico trasporto ionico

53
Calcola la TRAIETTORIA di un sistema molecolare = la configurazione molecolare in funzione del tempo, ovvero come variano nel tempo le posizioni, le velocità e le accelerazioni degli atomi della molecola. La traiettoria è generata da integrazioni simultanee dell’ equazione del moto di Newton Fi = mi ai per tutti gli atomi del sistema molecolare

54
Per calcolare una traiettoria c'è bisogno:
1. delle posizioni inziali ri : si ricavano da strutture sperimentali (cristallografia raggi X, NMR ecc.) o ottenute con modeling; 2. delle velocità iniziali vi : si ottengono dalla distribuzione delle velocità alla temperatura assegnata; 3. delle accelerazioni : sono determinate da una relazione che lega la forza che agisce su un atomo all'energia potenziale.
 o ottenute con modeling; 2. delle velocità iniziali vi : si ottengono dalla distribuzione delle velocità alla temperatura assegnata; 3. delle accelerazioni : sono determinate da una relazione che lega la forza che agisce su un atomo all energia potenziale.")
55
In pratica si considerano intervalli di integrazione finiti Dt
In pratica si considerano intervalli di integrazione finiti Dt. Dt tipicamente va da 0.1 a 10 fs per i sistemi molecolari una simulazione di 100 ps coinvolge intervalli di integrazione.

56
Protocollo di simulazione
Struttura iniziale rimuove interazioni di Van der Waals forti che porterebbero a distorsioni locali Minimizzazione dell’ energia nel caso si usi un solvente esplicito, aggiungere le molecole d’acqua Solvatazione della proteina Minimizzazione dell’ energia in presenza del solvente per equilibrarlo con la struttura si lancia la MD con velocità iniziali a bassa temperatura nuove velocità riassegnate periodicamente a T leggermente più alta e così via fino al raggiungimento della T di simulazione desiderata. Fase di riscaldamento la simulazione prosegue finchè sono stabili nel tempo la struttura, la pressione, la temperatura (si riscalano le velocità), l'energia Fase di equilibrazione Fase di simulazione vera e propria
, l energia. Fase di equilibrazione. Fase di simulazione vera e propria.")
57
Analisi dei risultati Analisi dei risultati
Campionamento periodico di coordinate (e velocità) Calcolo dell’energia potenziale media in funzione del tempo Calcolo della differenza con la struttura di partenza in funzione del tempo Calcolo della superficie accessibile al solvente e del raggio di girazione, in funzione del tempo Calcolo della struttura media
 Calcolo dell’energia potenziale media in funzione del tempo. Calcolo della differenza con la struttura di partenza in funzione del tempo. Calcolo della superficie accessibile al solvente e del raggio di girazione, in funzione del tempo. Calcolo della struttura media.")
58
Riconoscimento molecolare
E’ il punto di partenza per quasi tutti i processi biologici. Le molecole interagiscono in una maniera altamente specifica: modello CHIAVE-SERRATURA (Fisher e Ehrilch)
")
59
La complementarità geometrica e chimica fra piccole molecole biologiche (LIGANDI) e le strutture dei loro bersagli macromolecolari (RECETTORI) gioca un ruolo molto importante all’interno dei processi biologici.
 e le strutture dei loro bersagli macromolecolari (RECETTORI) gioca un ruolo molto importante all’interno dei processi biologici.")
60
La capacità di un recettore di agganciarsi al suo ligando selettivamente e con alta affinità è dovuta alla formazione di una serie di legami deboli (legami H, ionici, interazioni di Van der Waals) sommati ad interazioni favorevoli (es. idrofobiche). Gli atomi del sito di legame forniscono l’impalcatura necessaria per conferire alla superficie un dato contorno e particolari proprietà chimiche Variazioni anche piccole di tali amminoacidi spesso compromettono la funzionalità del recettore

61
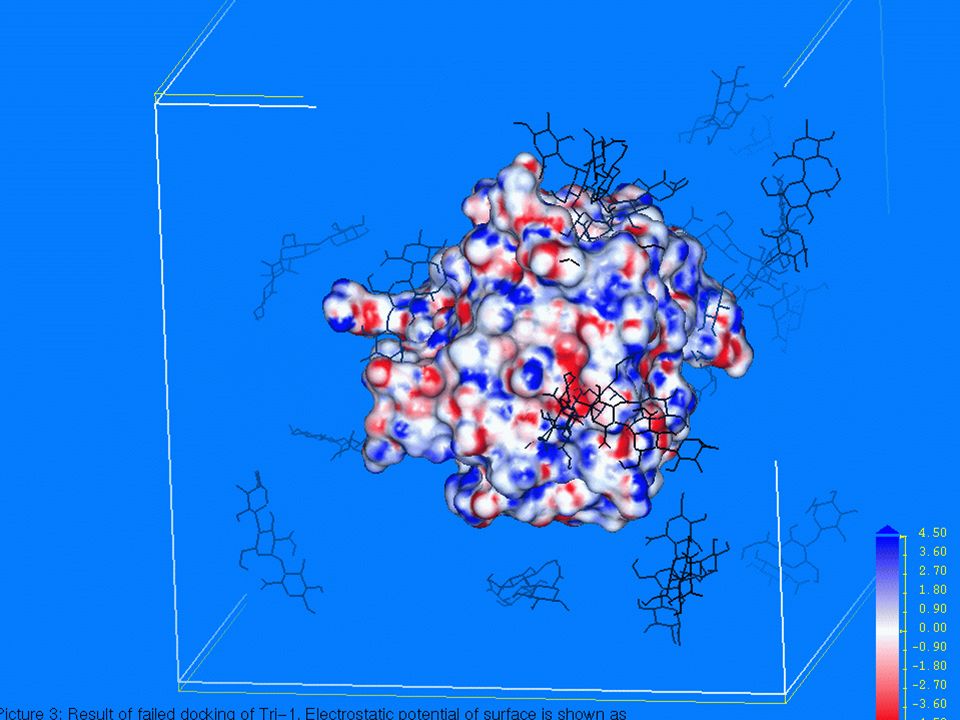
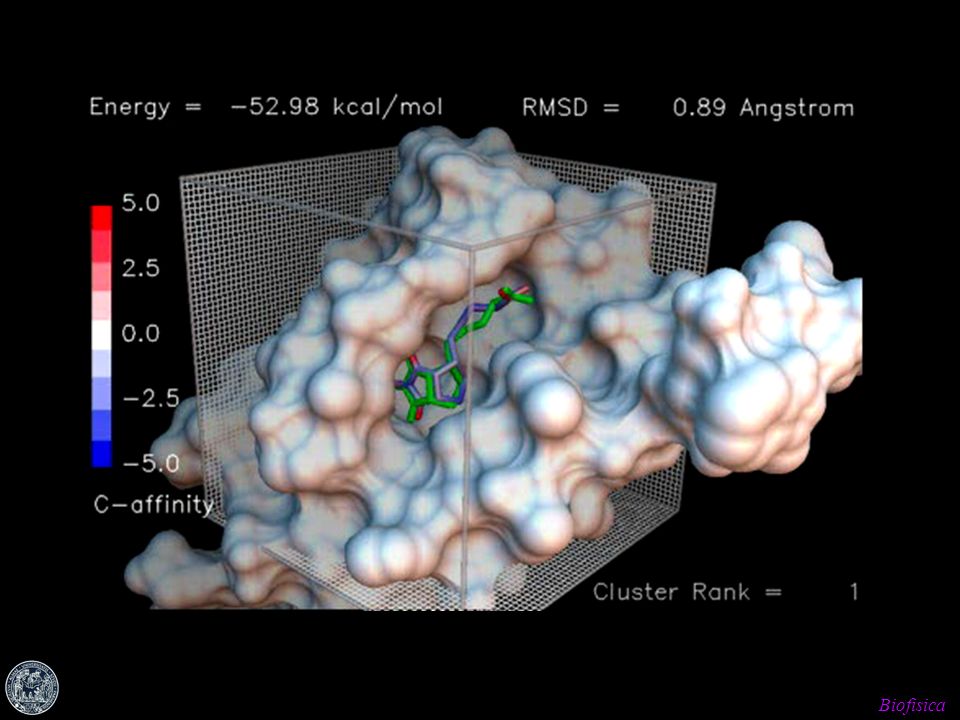
docking Docking Metodi computazionali per la predizione della struttura 3D di complessi proteina-ligando. Insieme di tecniche che consentono di simulare interazioni tra vari sistemi (sito attivo di un enzima e una nuova molecola di inibitore) e di valutare approssimativamente le nuove interazioni che si creano.
 e di valutare approssimativamente le nuove interazioni che si creano.")
62
In quest’ultimo caso, viene trovato il corretto modo di legame di un composto tramite il campionamento dello spazio conformazionale nel sito di legame, attraverso la valutazione di funzioni che stimano l’energia di ogni combinazione confomazionale ligando-recettore. Tali funzioni valutano fattori quali: Complementarità fra superficie Interazioni elettrostatiche e idrofobiche Energia libera di solvatazione

63
AUTODOCK, DOCK, FlexX, GOLD, GRID…
algoritmi AUTODOCK, DOCK, FlexX, GOLD, GRID… Algoritmi di docking. Posizionano ligandi - rigidi o flessibili - in siti di binding proteici generalmente rigidi. Il ligando durante il binding cambia la sua struttura tridimensionale in termini di angoli di torsione per trovare il miglior adattamento spaziale ed energetico nel sito di binding della proteina. Questa assunzione segue il modello “chiave-serratura” citato in precedenza. Prendendo in considerazione anche la flessibilità del recettore allora andrebbero considerati centinaia o anche migliaia di gradi di libertà.

64
scoperta di composti guida nuovi e innovativi
farmaci Sviluppo di farmaci Elemento chiave: scoperta di composti guida nuovi e innovativi Composto guida = composto che mostra affinità per un dato recettore, che ha attività biologica e che può essere strutturalmente modificato per migliorare la bioattività

65
tempi per lo sviluppo di un nuovo farmaco
Ricerca del composto guida (1-2 anni) Ottimizzazione del composto guida (1-2 anni) Saggi di attività in vitro e in vivo (1-2 anni) Test tossicologici (1-3 anni) Test per la sicurezza sull’uomo (1 anno) Test per l’efficacia sull’uomo (1-2 anni) Tempo totale per lo sviluppo di un nuovo farmaco: 6-12 anni Costo totale: circa $ è di grande importanza l’identificazione RAPIDA E AFFIDABILE di ligandi ad alta affinità
 Ottimizzazione del composto guida (1-2 anni) Saggi di attività in vitro e in vivo (1-2 anni) Test tossicologici (1-3 anni) Test per la sicurezza sull’uomo (1 anno) Test per l’efficacia sull’uomo (1-2 anni) Tempo totale per lo sviluppo di un nuovo farmaco: 6-12 anni. Costo totale: circa $ è di grande importanza l’identificazione RAPIDA E AFFIDABILE di ligandi ad alta affinità.")
66
Ricerca di nuovi composti guida
Screening sperimentale: test in vitro di grandi librerie di composti. Ignora, in genere, le proprietà strutturali del recettore Metodi computazionali detti Rational Design: si basano su informazioni strutturali del recettore e/o del ligando

67
Metodi computazionali
QSAR (Quantitaive Structure-Activity Relationship) Stabilisce una relazione tra la struttura molecolare e l’attività biologica di una serie di composti attivi. Predice la attività e la affinità di composti non noti dall’analisi delle similitudini e differenze strutturali con composti noti, fornendo anche informazioni sui requisiti strutturali del recettore. Structure Based Drug Design Struttura 3D del recettore NON nota Struttura 3D del recettore nota De Novo Design I nuovi composti vengono generati nel sito di legame a partire da atomi o frammenti preposizionati nel sito e che successivamente vengono trasformati in molecole intere da softwares specifici. Screening Virtuale Librerie di molecole (esistenti o ipotetiche) vengono analizzate cercando ligandi con caratteristiche in accordo con i requisiti del sito di legame Docking
 Stabilisce una relazione tra la struttura molecolare e l’attività biologica di una serie di composti attivi. Predice la attività e la affinità di composti non noti dall’analisi delle similitudini e differenze strutturali con composti noti, fornendo anche informazioni sui requisiti strutturali del recettore. Structure Based Drug Design. Struttura 3D del recettore NON nota. Struttura 3D del recettore nota. De Novo Design. I nuovi composti vengono generati nel sito di legame a partire da atomi o frammenti preposizionati nel sito e che successivamente vengono trasformati in molecole intere da softwares specifici. Screening Virtuale. Librerie di molecole (esistenti o ipotetiche) vengono analizzate cercando ligandi con caratteristiche in accordo con i requisiti del sito di legame. Docking.")
70
In conclusione... Le tecniche computazionali rappresentano uno strumento molto utile per: la modellazione di sistemi proteici la comprensione dei processi biologici la comprensione della relazione struttura-attività la scoperta e ottimizzazione dei composti guida farmacologici Vantaggio dal punto di vista biologico, chimico e farmaceutico, riducendo i tempi e completando e indirizzando le conoscenze sperimentali

Presentazioni simili






 – ridondante – strutture ottenute sperimentalmente via X-ray o NMR Il file PDB http://www.pdb.org Esempio:>")












>")