Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Espressione di un gene eucariotico

2
Il cloning di un gene richiede enzimi che tagliano il DNA in modo preciso e riproducibile

3
La scoperta degli enzimi di restrizione
Verso il 1950 si scoprì che alcuni batteri erano immuni a infezioni batteriofage, un fenomeno chiamato host-controlled restriction Presenti in tutti i batteri Più di 1200 sono stati caratterizzati Classificati in tre classi: tipo I, tipo II e tipo III Tipo I e III hanno un funzionamento complicato e non usati per l’ingegneria genetica (cloning) Tipo II sono usati come enzimi di restrizione per il cloning dei geni
 Tipo II sono usati come enzimi di restrizione per il cloning dei geni.")
4
Enzimi di restrizione BamHI EcoRI C G G A T C T G G C C A T T G A A C
Molti batteri producono sostanze che servono per difendersi dall’ invasione di DNA estraneo (per es: virale). Queste sostanze, dette enzimi di restrizione, attaccano il DNA e lo tagliano. Sono endonucleasi. Ciascuno di questi enzimi è in grado di riconoscere una specifica sequenza di basi (da 4 a 8) e di tagliare in corrispondenza di essa.
. Queste sostanze, dette. enzimi di restrizione, attaccano il DNA e lo tagliano. Sono endonucleasi. Ciascuno di questi enzimi è in grado di riconoscere una specifica. sequenza di basi (da 4 a 8) e di tagliare in corrispondenza di essa.")
5
Le sequenze di riconoscimento per alcuni frequenti enzimi di restrizione

6
Blunt and sticky ends

7
Enzimi di restrizione

8
Come si effettua una digestione in lab con gli enzimi di restrizione
Il tampone 10X: Mg2+ necessario per far funzionare efficientemente l’enzima DTT stabilizza l’enzima e impedisce la sua inattivazione MOLTO IMPORTANTE: avere le corrette concentrazioni di Mg2+ e NaCl per impedire scarsa attività e specificità dell’enzima

9
Come si effettua una digestione in lab con gli enzimi di restrizione
L’enzima di restrizione: 1 unità di enzima è definita come la quantità necessaria per tagliare 1 mg di DNA in 1 ora, quindi… Abbiamo bisogno di 2 unità per 2 mg di l DNA ed, essendo BglII fornita a concentrazione corrispondente a 4 unità/ml, aggiungiamo 0.5 ul per tagliare 2 mg di DNA in 1 ora Per bloccare la reazione dopo 1 ora si può “uccidere” l’enzima in vari modi, per esempio una breve incubazione a 70°C o l’aggiunta di EDTA, etc.

10
Come si effettua una digestione in lab con gli enzimi di restrizione
Consideriamo di dover digerire un l DNA (2 mg/ml) con BglII:
 con BglII:")
11
Espressione di un gene eucariotico

12
Cos’è la PCR (Polymerase Chain Reaction) ?
E’ una tecnica per l’amplificazione “in vitro” di specifiche sequenze di DNA, attraverso la simultanea estensione di filamenti complementari di DNA. Il metodo fu inventato da Mullis e colleghi della Cetus Corporation nel 1987, sfruttando una reazione naturale compiuta dalla DNA polimerasi 5’ DNA polimerasi primer 3’ dNTP mix+ Mg2+ 5’ 3’ 5’ 3’ 3’ 5’ La PCR utilizza lo stesso principio,utilizzando 2 primers, ognuno complementare a filamenti opposti di DNA, denaturati per riscaldamento

13
L’invenzione della PCR
Ideata da Kary Mullis nel 1983 La prima pubblicazione è apparsa nel 1985 Premio Nobel per la chimica nel 1995 Libro del 1998 ("Ballando nudi nel campo della mente. Le idee (e le avventure) del più eccentrico tra gli scienziati moderni")
 del più eccentrico tra gli scienziati moderni )")
14
PCR Denaturazione DNA Raffreddamento necessario per il processo di ibridizzazione (fase di annealing) Sintesi della regione da amplificare a opera della Taq polimerasi (fase di estensione)
")
15
Schema di una reazione PCR
Usando 2 primers che si appaiano a filamenti complementari di DNA, vengono sintetizzati 2 nuovi filamenti. Se il processo viene ripetuto, sia il campione di DNA che i nuovi filamenti possono servire come templati, portando ad un aumento esponenziale di prodotto che termina con le posizioni definite dai primers. Prodotti con il primer solo ad un’estremità e quindi di lunghezza indeterminata aumentano in maniera lineare durante il processo e, insieme al DNA di partenza, formano solo una piccola frazione del prodotto della PCR.

16
PCR Cycles

17
Componenti e fasi della reazione
Miscela di reazione: l 1) Tampone dNTPs Primers Templato (DNA a doppio filamento) DNA polimerasi termostabile Fasi della reazione 1) Separazione o denaturazione (30” a 94°C per frammenti 1 Kb) I 2 filamenti della molecola di DNA vengono separati portando la miscela a 94°C per 15”-30”. Ibridizzazione La miscela viene raffreddata a T Tm – 2/3°C, per 30”-60” perché ciascun primer si appaia con un filamento di DNA. Estensione La soluzione viene riscaldata a 72°C, temperatura ottimale per l’attività della polimerasi, per 1’-2’ per amplificare frammenti di 1 Kb. 1° ciclo 2° ciclo 94°C 94°C initial melt melt 72 °C melt 72 °C 50-60 °C estensione 50-60 °C estensione ibridizzazione ibridizzazione
 Tampone. dNTPs. Primers. Templato (DNA a doppio filamento) DNA polimerasi termostabile. Fasi della reazione. 1) Separazione o denaturazione (30 a 94°C per frammenti 1 Kb) I 2 filamenti della molecola di DNA vengono separati portando la miscela a 94°C per Ibridizzazione. La miscela viene raffreddata a T Tm – 2/3°C, per perché ciascun primer si appaia con un filamento di DNA. Estensione. La soluzione viene riscaldata a 72°C, temperatura ottimale per l’attività della polimerasi, per 1’-2’ per amplificare frammenti di 1 Kb. 1° ciclo. 2° ciclo. 94°C. 94°C. initial melt. melt. 72 °C. melt. 72 °C °C. estensione °C. estensione. ibridizzazione. ibridizzazione.")
18
Come disegnare i primers ?
I primers devono corrispondere alle sequenze di basi che fiancheggiano il gene target nel DNA templato Primer deve essere complementare (non uguale!) allo strand da amplificare nel DNA templato Frammento di DNA da amplificare ideale lunghezza di 3Kb. Frammenti più lunghi di 10 Kb sono amplificati correttamente ma quando più lunghi la reazione è meno efficiente
 allo strand da amplificare nel DNA templato. Frammento di DNA da amplificare ideale lunghezza di 3Kb. Frammenti più lunghi di 10 Kb sono amplificati correttamente ma quando più lunghi la reazione è meno efficiente.")
19
Quanto lunghi devono essere i primers ?
Se troppo corti: Nel genoma umano per un primer 8-mer si ha circa possibili siti di attacco tra i kb di sequenza nucleotidica del genoma umano Se primer 17-mer otteniamo una solo specifico possibile sito di attacco nel genoma umano Se troppo lunghi: La lunghezza del primer influenza la velocità di ibridizzazione al templato, cioè primer lunghi si legano a velocità più lenta. Quindi risulta una bassa efficienza della PCR. In pratica, primer più lunghi di 30-mer si usano raramente.

20
Quale è la temperatura adatta da usare nella PCR ?
La temperature di ibridizzazione o annealing influenza la specificità della reazione Melting temperature Tm : temperatura alla quale il corretto ibrido si dissocia (“melts”) Quindi la temperatura ideale deve essere bassa abbastanza da permettere la ibridizzazione tra il primer e il DNA templato ma alta abbastanza da impedire una ibridizzazione mismatched.
 Quindi la temperatura ideale deve essere bassa abbastanza da permettere la ibridizzazione tra il primer e il DNA templato ma alta abbastanza da impedire una ibridizzazione mismatched.")
21
Tm = (4 X [G+C]) + (2 X [A+T]) °C
Come calcolare Tm ? Sperimentalmente può essere determinata, ma di solito si utilizza la seguente formula: Tm = (4 X [G+C]) + (2 X [A+T]) °C Quindi la temperatura di annealing per la PCR è calcolata 1-2°C sotto il Tm. IMPORTANTE: I due primers devono essere disegnati in modo da avere la stessa Tm
![Tm = (4 X [G+C]) + (2 X [A+T]) °C](http://slideplayer.it/slide/2582385/9/images/21/Tm+%3D+%284+X+%5BG%2BC%5D%29+%2B+%282+X+%5BA%2BT%5D%29+%C2%B0C.jpg "Come calcolare Tm Sperimentalmente può essere determinata, ma di solito si utilizza la seguente formula: Tm = (4 X [G+C]) + (2 X [A+T]) °C. Quindi la temperatura di annealing per la PCR è calcolata 1-2°C sotto il Tm. IMPORTANTE: I due primers devono essere disegnati in modo da avere la stessa Tm.")
22
Componenti e condizioni di reazione
Soluzione tampone -10 mM Tris pH 8.4 -50 mM KCl -1.5 mM MgCl2 -0.01 % Detergente non ionico (Tween 20 o Triton X-100) La concentrazione di KCl e MgCl2 può essere variata fino a trovare le concentrazioni ottimali per la reazione dNTPs=deossinucleosidi trifosfato Concentrazione finale 200 M ognuno (soluzione stock: 10 mM ognuno) Target DNA o templato E’ essenziale che sia intatto nella zona da amplificare e privo di sostanze che possano inibire la reazione della polimerasi Prima dell’inizio dei cicli di reazione si denatura completamente il templato scaldando per 5’ a °C per evitare estensioni aspecifiche. La Taq polimerasi viene aggiunta dopo questo step. Il templato si usa in quantità di ng, in 50 l di volume di reazione.
 La concentrazione di KCl e MgCl2 può essere variata fino a trovare le concentrazioni ottimali per la reazione. dNTPs=deossinucleosidi trifosfato. Concentrazione finale 200 M ognuno (soluzione stock: 10 mM ognuno) Target DNA o templato. E’ essenziale che sia intatto nella zona da amplificare e privo di sostanze che possano inibire la reazione della polimerasi. Prima dell’inizio dei cicli di reazione si denatura completamente il templato scaldando per 5’ a °C per evitare estensioni aspecifiche. La Taq polimerasi viene aggiunta dopo questo step. Il templato si usa in quantità di ng, in 50 l di volume di reazione.")
23
Componenti e condizioni di reazione
Primers La sequenza dei primers deve essere complementari alle estremità del frammento da amplificare. -Non deve avere omologie con altre zone del templato -I 2 primers hanno lunghezza di basi e simile contenuto di (G+C), minima struttura secondaria, e bassa complementarità l’uno con l’altro Tm= 4(G+C) +2(A+T) -Tm= temperatura di melting -Rapporto conc. molare primers/templato = 107. -Concentrazione primers pmoli in 50 l DNA polimerasi DNA polimerasi termostabili Thermus aquaticus Thermus thermophilus Bacillus stereothermophilus Thermococcus litoralis Richiede ioni Mg2+ per l’attività polimerasica. Si usa in concentrazione 1-2 U in 50 l Numero di cicli 25-30 cicli sono sufficienti per ottenere sufficiente prodotto.
, minima struttura secondaria, e bassa complementarità l’uno con l’altro. Tm= 4(G+C) +2(A+T) -Tm= temperatura di melting. -Rapporto conc. molare primers/templato = Concentrazione primers pmoli in 50 l. DNA polimerasi. DNA polimerasi termostabili. Thermus aquaticus. Thermus thermophilus. Bacillus stereothermophilus. Thermococcus litoralis. Richiede ioni Mg2+ per l’attività polimerasica. Si usa in concentrazione 1-2 U in 50 l. Numero di cicli cicli sono sufficienti per ottenere sufficiente prodotto.")
24
Come analizzare i prodotti della PCR?
Agarose gel elettroforesi Diretta analisi di sequenza del prodotto della PCR Analisi che coinvolgono il cloning del prodotto di PCR

25
Agarose gel elettroforesi
Miscela di prodotti Amplificazione di dimensioni sbagliate Nessuna amplificazione

26
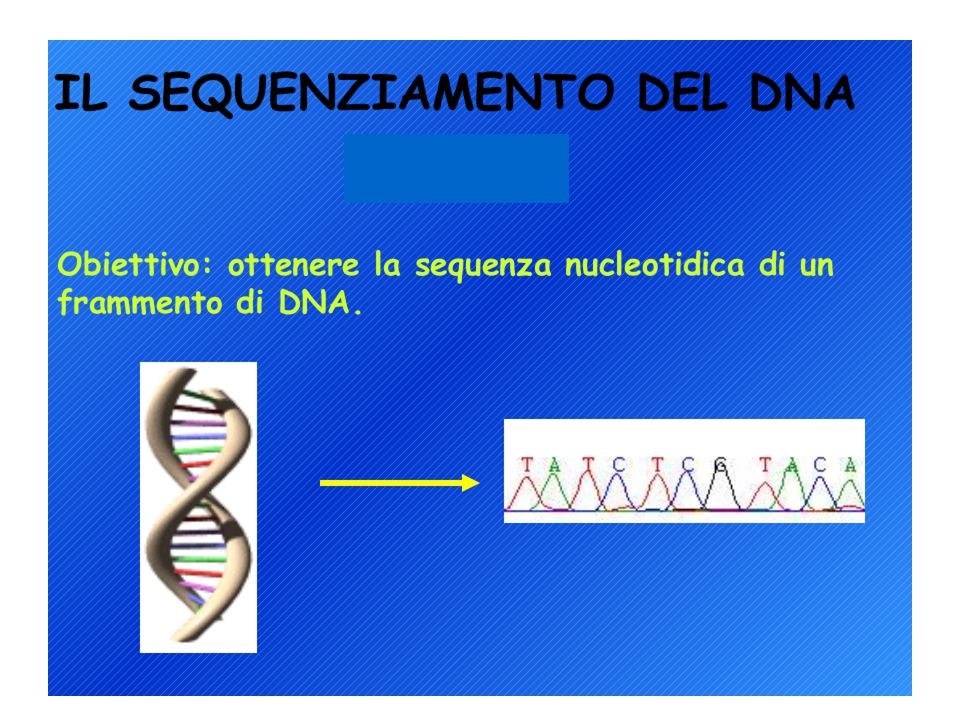
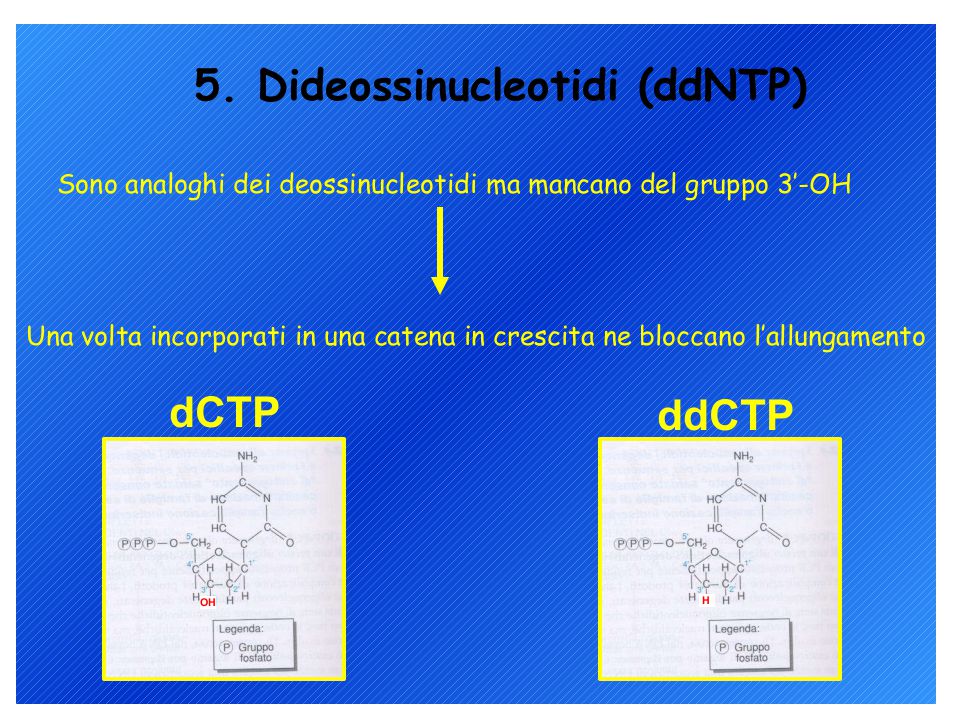
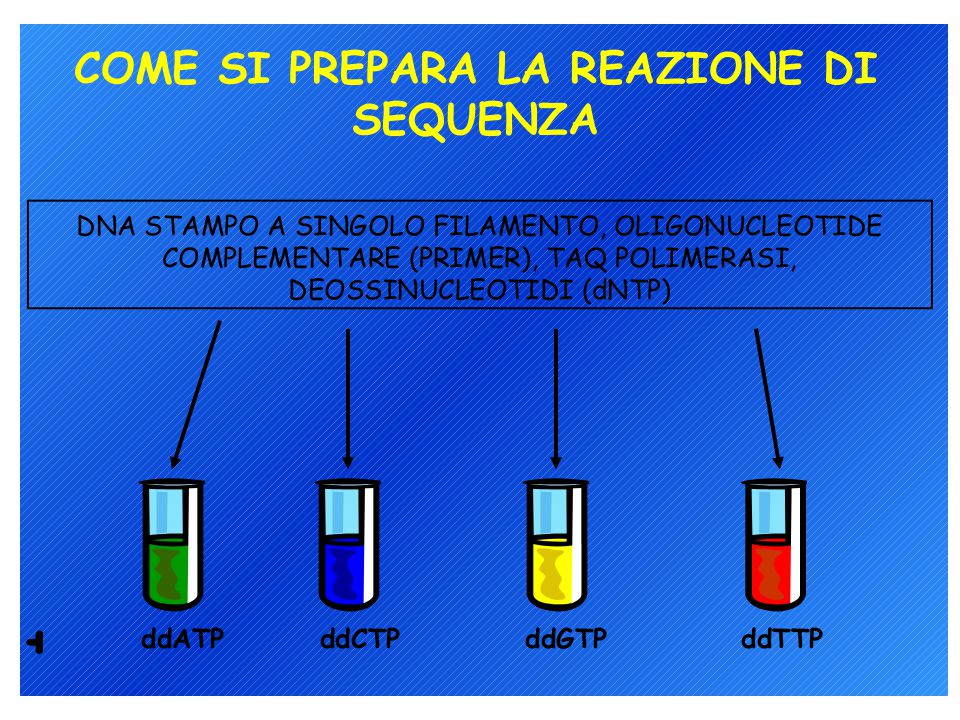
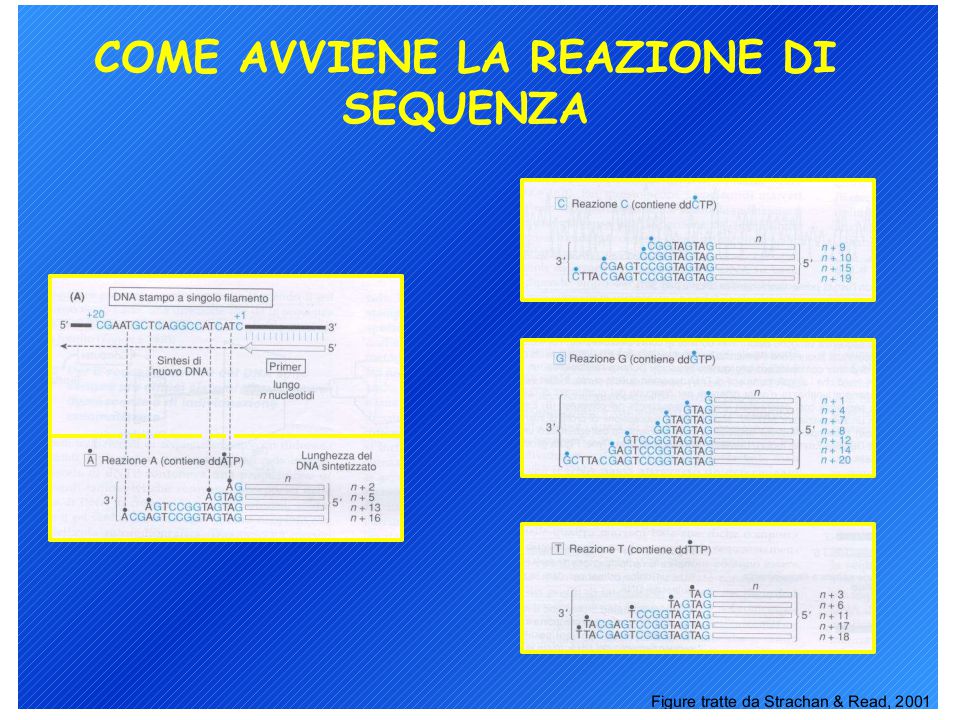
Analisi diretta dei prodotti di PCR
Metodi di sequenziamento del DNA: 1. Metodo di Sanger (metodo enzimatico) 2. Metodo Maxam-Gilbert (metodo chimico) L’analisi può richiedere un single-stranded DNA come materiale di partenza ma il prodotto di PCR è double-stranded quindi…. Biotina-labelled primer affine per proteina avidina
 2. Metodo Maxam-Gilbert (metodo chimico) L’analisi può richiedere un single-stranded DNA come materiale di partenza ma il prodotto di PCR è double-stranded quindi…. Biotina-labelled primer affine per proteina avidina.")
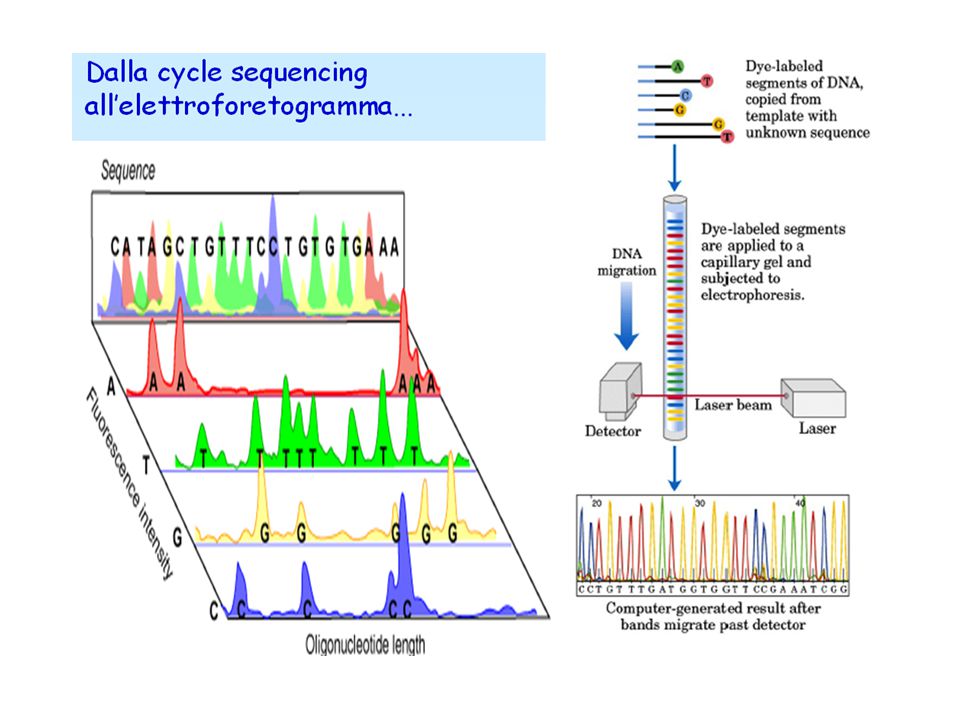
38
Con l’avvento dei sequenziatori automatici, si è iniziato a fare tutto in un’unica miscela di reazione, etichettando ogni base azotata con una molecola fluorescente di colore diverso; inoltre, i frammenti si separano in tubi capillari, con un lettore ottico che registra il colore emesso dal DNA al suo passaggio.

40
Cloning dei prodotti di PCR
Taq polymerasi tende ad aggiungere un nucleotide addizionale, di solito adenosina alla fine di ciascuno strand: Il prodotto di PCR non è blunt-ended Esonucleasi enzimi possono rimuovere la A, ma bassa selettività quindi…

41
Prima soluzione: Il vettore si prepara tagliandolo ad un sito blunt-ended e poi trattandolo con Taq polimerasi in presenza di dTTP In assenza di primer la polimerasi può solo aggiungere una T alla parte 3’

42
Seconda soluzione (la più usata):
Disegnare primer che contengono siti di restrizione nel DNA templato: L’approccio non è limitato ai casi in cui i siti di restrizione sono presenti nel DNA templato Di solito infatti i siti di restrizione sono aggiunti alle sequenze dei primers come corti segmenti in posizione 5’:

Presentazioni simili