Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Determinazione teorica della geometria molecolare Benché le moderne tecniche NMR possono dare alcune indicazioni sulle distanze interatomiche in campioni liquidi, le informazioni geometriche per molecole grandi sono attualmente derivate principalmente da dati cristallografici. Ciò nonostante queste informazioni riguardano solo i cristalli e non sono automaticamente le stesse, per esempio, delle specie reagenti in mezzi biologici. Inoltre questi dati non possono essere ottenuti per campioni che non danno buoni cristalli, per strutture ipotetiche o per molecole non ancora sintetizzate. Infine se i dati dei raggi X danno la geometria della forma più stabile, non danno alcuna informazione sull’energia o sulla possibile esistenza di altre strutture a bassa energia. Per avere questo tipo di informazioni bisogna dunque ricorrere a previsioni di tipo computazionale.

2
Determinazione teorica della geometria molecolare Il sistema ad energia più bassa è quello più stabile. Trovare la geometria di una molecola significa trovare la struttura con l'energia minore. Occorre quindi un metodo per calcolare l’energia di una molecola (o di un sistema di molecole), note le posizioni degli atomi che la compongono: Esistono due modelli per valutare l’energia di una molecola: 1)Meccanica quantistica 2)Meccanica molecolare (basato sulla meccanica classica)
, note le posizioni degli atomi che la compongono: Esistono due modelli per valutare l’energia di una molecola: 1)Meccanica quantistica 2)Meccanica molecolare (basato sulla meccanica classica).")
3
Si applica alle particelle macroscopiche ma perde di validità per quelle microscopiche (fotoni, elettroni, nuclei(?)) Le particelle sono descritte dalle loro posizioni istantanee r (t) e dalle velocità v = dr / dt La posizione istantanea r (t) di una particella definisce la sua traiettoria. r (t) è un vettore (x(t),y(t),z(t)) e definisce come evolve nel tempo la posizione della particella in un sistema di coordinate cartesiane (x,y,z) Meccanica classica
 è un vettore (x(t),y(t),z(t)) e definisce come evolve nel tempo la posizione della particella in un sistema di coordinate cartesiane (x,y,z) Meccanica classica.")
4
Energia totale di un sistema di particelle in meccanica classica Nel caso specifico l’energia totale di un sistema di particelle può essere considerata come la somma di energia cinetica e energia potenziale: E tot = E kin + V Es: coppia di particelle cariche di massa m 1 e m 2 che si muovono con velocità v 1 e v 2 e poste alla distanza r: E kin = ½ m 1 v 1 2 + ½ m 2 v 2 2 L’energia potenziale è di tipo coulombiano:

5
Energia totale di un sistema di particelle in meccanica quantistica All’inizio del 1900 varie osservazioni sperimentali resero evidente che la meccanica classica non si applica a fenomeni molecolari ed atomici. In particolare risultò che particelle microscopiche hanno una natura ondulatoria e questo poneva dei limiti alla determinazione simultanea della loro posizione e velocità. Lunghezza d’onda di de Broglie: = h/mv Principio di indeterminazione di Heisenberg: ( x)(m v) = h/4
(m v) = h/4 .")
6
Energia totale di un sistema di particelle in meccanica quantistica Per un sistema di particelle per le quali vale la meccanica quantistica, tutte le quantità osservabili del sistema possono essere calcolate se si conosce la funzione d’onda del sistema. Ad ogni particella quantistica può infatti essere Ψ (x,y,z) tale che il suo quadrato | Ψ (x,y,z)| 2 dà la probabilità di trovare la particella nel punto dello spazio di coordinate (x,y,z). Ad esempio per un sistema di due particelle quantistiche la funzione d’onda dipenderà dalle coordinate (x1,y1,z1) della prima particella e (x2,y2,z2) della seconda particella. Il quadrato della funzione d’onda |Ψ(x1,y1,z1,x2,y2,z2)| 2 ci darà la probabilità di trovare la particella 1 nel punto (x1,y1,z1) e contemporaneamente la particella 2 nel punto (x2,y2,z2) Ψ (x,y,z)è una funzione d’onda che descrive la particella, ma in sé non ha un significato fisico, è solo un artificio matematico. Ψ (x,y,z) è una funzione d’onda che descrive la particella, ma in sé non ha un significato fisico, è solo un artificio matematico.
 tale che il suo quadrato | Ψ (x,y,z)| 2 dà la probabilità di trovare la particella nel punto dello spazio di coordinate (x,y,z). Ad esempio per un sistema di due particelle quantistiche la funzione d’onda dipenderà dalle coordinate (x1,y1,z1) della prima particella e (x2,y2,z2) della seconda particella. Il quadrato della funzione d’onda |Ψ(x1,y1,z1,x2,y2,z2)| 2 ci darà la probabilità di trovare la particella 1 nel punto (x1,y1,z1) e contemporaneamente la particella 2 nel punto (x2,y2,z2) Ψ (x,y,z)è una funzione d’onda che descrive la particella, ma in sé non ha un significato fisico, è solo un artificio matematico. Ψ (x,y,z) è una funzione d’onda che descrive la particella, ma in sé non ha un significato fisico, è solo un artificio matematico..")
7
Energia totale di un sistema di particelle in meccanica quantistica Per ottenere le quantità osservabili del sistema basta applicare alla funzione d’onda un operatore (= simbolo che applicato ad una certa funzione dice ciò che essa deve diventare) appropriato. Esempio: Operatore energia cinetica: - (h 2 /8 2 m) 2 dove 2 = 2 / x 2 + 2 / y 2 + 2 / z 2 Laplaciano Operatore energia potenziale: V(x,y,z) Operatore momento di dipolo:
 2 dove 2 = 2 / x 2 + 2 / y 2 + 2 / z 2 Laplaciano Operatore energia potenziale: V(x,y,z) Operatore momento di dipolo:.")
8
Energia totale di un sistema di particelle in meccanica quantistica Per calcolare quindi l’energia totale del sistema dovrò sommare l’energia cinetica con quella potenziale, avrò quindi l’operatore: Operatore energia totale = Hamiltoniano = H Ĥ =E kin +V= - (h 2 /8 2 m) 2 + V Questo vuol dire che se applico H alla funzione d’onda del sistema ho l’energia totale del sistema stesso: Ĥ = E oppure: [ - (h 2 /8 2 m) 2 - V(x,y,z) ] ( x,y,z ) = E ( x,y,z ) Questa è l’equazione di Schrödinger. Essa è particolarmente importante, perchè si usa anche per ricavare la funzione d’onda.
![Energia totale di un sistema di particelle in meccanica quantistica Per calcolare quindi l’energia totale del sistema dovrò sommare l’energia cinetica con quella potenziale, avrò quindi l’operatore: Operatore energia totale = Hamiltoniano = H Ĥ =E kin +V= - (h 2 /8 2 m) 2 + V Questo vuol dire che se applico H alla funzione d’onda del sistema ho l’energia totale del sistema stesso: Ĥ = E oppure: [ - (h 2 /8 2 m) 2 - V(x,y,z) ] ( x,y,z ) = E ( x,y,z ) Questa è l’equazione di Schrödinger.](http://images.slideplayer.it/11/3061869/slides/slide_8.jpg "Essa è particolarmente importante, perchè si usa anche per ricavare la funzione d’onda..")
9
Energia totale di un sistema di particelle in meccanica quantistica: atomo di idrogeno Il problema è che tale equazione (per i sistemi che ci interessano) è risolvibile analiticamente solo per l’atomo di idrogeno (o per ioni con un solo elettrone). Tale sistema è costituito da un elettrone che si muove nel potenziale elettrostatico del nucleo fisso di carica +e. Il potenziale che agisce sull’elettrone è quello elettrostatico V= -e 2 /r e l’equazione di Schrödinger diventa: [ - (h 2 /8 2 m) 2 - e 2 / r ] ( x,y,z ) = E ( x,y,z ). n,l,m (r, , ) = R n,l (r )Y l,m ( , ) Tale equazione è risolvibile analiticamente in coordinate polari sferiche
 2 - e 2 / r ] ( x,y,z ) = E ( x,y,z ). n,l,m (r, , ) = R n,l (r )Y l,m ( , ) Tale equazione è risolvibile analiticamente in coordinate polari sferiche.")
10
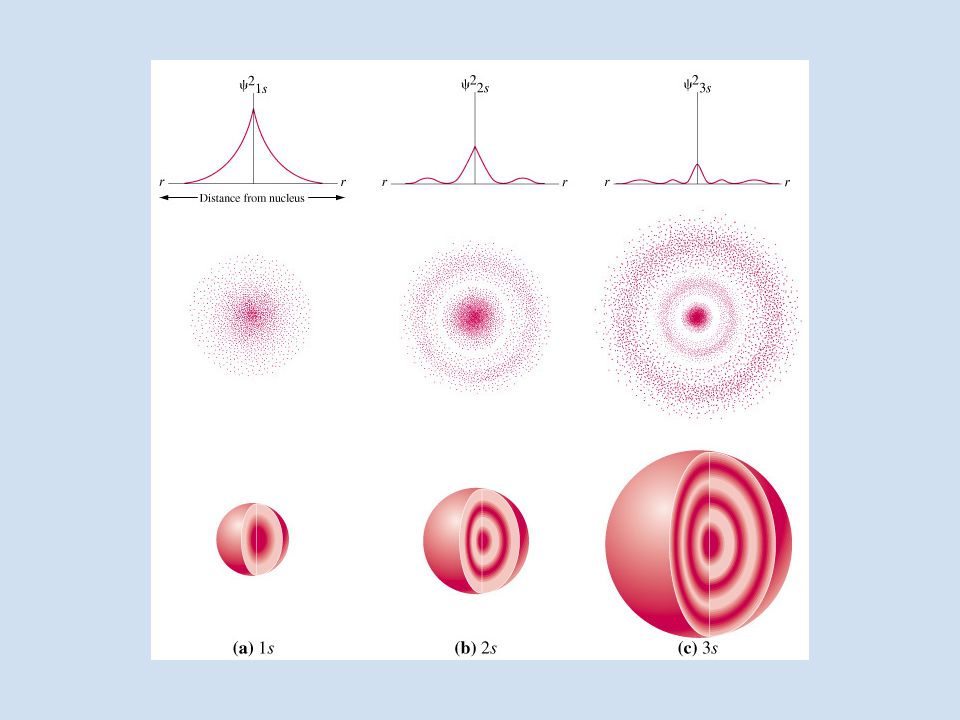
Le soluzioni sono infinite e dipendono dai numeri quantici n,l,m che sono semplicemente numeri interi o relativi che derivano dalla soluzione matematica dell’equazione di Schrödinger e classificano le possibili soluzioni. Ciascuna di queste soluzioni corrisponde ad un orbitale atomico e costituisce uno stato del sistema n=1, l=0, m=0 100 (r, , ) orbitale 1s n=2, l=0, m=0 200 (r, , ) orbitale 2s n=2, l=1, m= -1,0,+1 21-1 (r, , ) 210 (r, , ) 211 (r, , ) i tre orbitali 2p Significato fisico | 100 (r, , )| 2 probabilità che l’elettrone nell’orbitale 1s sia nel punto di coordinate (r, , ) attorno al nucleo | 200 (r, , )| 2 probabilità che l’elettrone nell’orbitale 2s sia nel punto di coordinate (r, , ) attorno al nucleo ……………….. Le energie dei vari stati dipendono solo da n sono dati dalla stessa equazione di Bohr E n = - R H / n 2 R H costante di Rydberg Unità di misura Hartree. Questa è un’energia assoluta, è cioè l’energia liberata per portare il nucleo dell’idrogeno e un elettrone da distanza infinita alla distanza d’interazione.
 orbitale 1s n=2, l=0, m=0 200 (r, , ) orbitale 2s n=2, l=1, m= -1,0,+1 21-1 (r, , ) 210 (r, , ) 211 (r, , ) i tre orbitali 2p Significato fisico | 100 (r, , )| 2 probabilità che l’elettrone nell’orbitale 1s sia nel punto di coordinate (r, , ) attorno al nucleo | 200 (r, , )| 2 probabilità che l’elettrone nell’orbitale 2s sia nel punto di coordinate (r, , ) attorno al nucleo ……………….. Le energie dei vari stati dipendono solo da n sono dati dalla stessa equazione di Bohr E n = - R H / n 2 R H costante di Rydberg Unità di misura Hartree. Questa è un’energia assoluta, è cioè l’energia liberata per portare il nucleo dell’idrogeno e un elettrone da distanza infinita alla distanza d’interazione..")
11
n,l,m (r, , ) = R n,l (r )Y l,m ( , )
 = R n,l (r )Y l,m ( , )")
13
2,1,0 (r, , ) = R 2,1 (r )Y 1,0 ( , )= R 2,1 (r )·( 3/ )cos Orbitale 2p
 = R 2,1 (r )Y 1,0 ( , )= R 2,1 (r )·( 3/ )cos Orbitale 2p")
14
Energia totale di un sistema di particelle in meccanica quantistica: atomi polielettronici Un atomo polielettronico è costituito da N elettroni che si muovono nel potenziale elettrostatico del nucleo fisso di carica +Ze. L’equazione di Schrödinger diventa: N N N N [ - (h 2 /8 2 m) i 2 - Z e 2 / r i + e 2 / r i,j ] = E i i i < j j La funzione d’onda dipende ora dalle coordinate degli N elettroni (cioè da 3N variabili) ( x 1,y 1,z 1, x 2,y 2,z 2,….,x N,y N,z N ) Questa equazione non è risolvibile analiticamente, bisogna ricorrere a metodi numerici (al computer).
 i 2 - Z e 2 / r i + e 2 / r i,j ] = E i i i < j j La funzione d’onda dipende ora dalle coordinate degli N elettroni (cioè da 3N variabili) ( x 1,y 1,z 1, x 2,y 2,z 2,….,x N,y N,z N ) Questa equazione non è risolvibile analiticamente, bisogna ricorrere a metodi numerici (al computer)..")
15
Energia totale di un sistema di particelle in meccanica quantistica: atomi polielettronici Uno dei metodi più usati è il metodo di Hartree o metodo del campo auto- consistente (self-consistent field). In questo metodo si trascurano i termini di repulsione interelettronici e 2 / r i,j e si fa l’approssimazione che ogni elettrone si muove nel campo efficace dovuto alla media del nucleo e degli altri elettroni. In tal caso l’equazione di Schrödinger diventa [ - (h 2 /8 2 m) i 2 - Z* e 2 / r i ] = E Z* carica nucleare efficace i i Ĥ(i) = E ed equivale matematicamente a N equazioni del tipo per l’atomo di idrogeno, con hamiltoniano Ĥ(i). Si dimostra che le soluzioni hanno la forma di un prodotto di N funzioni d’onda monoelettroniche identiche a quelle viste per l’atomo di idrogeno (x 1,y 1,z 1, x 2,y 2,z 2,….,x N,y N,z N ) = 1 (x 1,y 1,z 1 )· 2 (x 2,y 2,z 2,) · · · N (x N,y N,z N )
 i 2 - Z* e 2 / r i ] = E Z* carica nucleare efficace i i Ĥ(i) = E ed equivale matematicamente a N equazioni del tipo per l’atomo di idrogeno, con hamiltoniano Ĥ(i). Si dimostra che le soluzioni hanno la forma di un prodotto di N funzioni d’onda monoelettroniche identiche a quelle viste per l’atomo di idrogeno (x 1,y 1,z 1, x 2,y 2,z 2,….,x N,y N,z N ) = 1 (x 1,y 1,z 1 )· 2 (x 2,y 2,z 2,) · · · N (x N,y N,z N ).")
16
Energia totale di un sistema di particelle in meccanica quantistica: atomi polielettronici [ - (h 2 /8 2 m) i 2 - Z* e 2 / r i ] = E Z* carica nucleare efficace i i In questa equazione, in realtà Z* per ogni elettrone dipende dalla funzione d’onda degli altri elettroni, quindi le equazioni devono essere risolte contemporaneamente. Per fare questo si usa il metodo self-consistent field. Si parte da una funzione d’onda tentativo e si inserisce nell’equazione per ottenere il valore del campo, dell’energia e della nuova funzione d’onda. Si ripete (itera) il procedimento fino a che si ottiene sempre lo stesso insieme di funzioni d’onda e d energia.
![Energia totale di un sistema di particelle in meccanica quantistica: atomi polielettronici [ - (h 2 /8 2 m) i 2 - Z* e 2 / r i ] = E Z* carica nucleare efficace i i In questa equazione, in realtà Z* per ogni elettrone dipende dalla funzione d’onda degli altri elettroni, quindi le equazioni devono essere risolte contemporaneamente.](http://images.slideplayer.it/11/3061869/slides/slide_16.jpg "Per fare questo si usa il metodo self-consistent field. Si parte da una funzione d’onda tentativo e si inserisce nell’equazione per ottenere il valore del campo, dell’energia e della nuova funzione d’onda. Si ripete (itera) il procedimento fino a che si ottiene sempre lo stesso insieme di funzioni d’onda e d energia..")
17
Metodo self-consistent per la soluzione di equazioni di secondo grado. Supponiamo di dover risolvere l’equazione x 2 -8x+14=0 Soluzione analitica 2,5857864 5,4142136

18
Metodo self-consistent per la soluzione di equazioni di secondo grado. Soluzione numerica autoconsistente x 2 -8x+14=0 Riscrivo l’equazione come: Prendo una soluzione tentativo, per esempio x=1,000000000 1° ciclo metto la soluzione nell’equazione e ottengo : x=1,875000000 2° ciclo metto questa soluzione di nuovo nell’equazione e ottengo: x=2,189453125 3° ciclo

19
Metodo self-consistent per la soluzione di equazioni di secondo grado. Ottengo così questa serie di cicli: x=1,000000000 1° ciclo x=1,875000000 2° ciclo x=2,189453125 3° ciclo x=2,349213123 x=2,439850287 x=2,494108678 x=2,527572262 x=2,548577692 x=2,561906032 x=2,570420314 x=2,575882570 x=2,579396379 x=2,581660710 x=2,583121503 x=2,584064587 x=2,584673724 x=2,585067280 x=2,585321607 x=2,585485976 x=2,585592217 x=2,585660889 x=2,585705279 x=2,585733974 x=2,585752523.... x=2.585780515 Quando ci fermiamo?

20
Conviene ovviamente scrivere ciascuna funzione d’onda monoelettronica in funzione delle coordinate sferiche: i (x i,y i,z i ) = i n,l,m (r i, i, i ) = R in,l (r i )Y i l,m ( , ) In questa approssimazione la funzione d’onda polielettronica ha formalmente la stessa espressione della configurazione elettronica considerata per quell’atomo. Ad esempio per l’atomo di boro, Z=5, la configurazione elettronica 1s 2 2s 2 p 1 corrisponde alla funzione d’onda ( x 1,y 1,z 1,x 2,y 2,z 2,x 3,y 3,z 3,x 4,y 4,z 4,x 5,y 5,z 5 ) = 1s (x 1,y 1,z 1 )· 1s (x 2,y 2,z 2,)· 2s (x 3,y 3,z 3 )· 2s (x 2,y 2,z 2,)· 2p (x 3,y 3,z 3 ) in cui 1s è la 100 con n=1, l=0, m=0, 1s è la 200 con n=2, l=0, m=0, e così via. Per un dato atomo le soluzioni possibili sono infinite e corrispondono alle possibili configurazioni elettroniche cioè ai possibili modi (infiniti) di distribuire gli elettroni sui vari orbitali Quello più in basso in energia è lo stato fondamentale tutti gli altri sono detti stati eccitati. Anche in questo caso l’energia è un’energia assoluta: corrisponde a portare gli elettroni e il nucleo da distanze infinite alla posizione considerata.
 = 1s (x 1,y 1,z 1 )· 1s (x 2,y 2,z 2,)· 2s (x 3,y 3,z 3 )· 2s (x 2,y 2,z 2,)· 2p (x 3,y 3,z 3 ) in cui 1s è la 100 con n=1, l=0, m=0, 1s è la 200 con n=2, l=0, m=0, e così via. Per un dato atomo le soluzioni possibili sono infinite e corrispondono alle possibili configurazioni elettroniche cioè ai possibili modi (infiniti) di distribuire gli elettroni sui vari orbitali Quello più in basso in energia è lo stato fondamentale tutti gli altri sono detti stati eccitati. Anche in questo caso l’energia è un’energia assoluta: corrisponde a portare gli elettroni e il nucleo da distanze infinite alla posizione considerata..")
21
Energia totale di un sistema di particelle in meccanica quantistica: molecole Nel caso di una molecola la situazione è molto più complessa, principalmente per due ragioni 1) La funzione d’onda dipende anche dalle coordinate nucleari oltre che da quelle elettroniche = (nuclei,elettroni) Ad esempio per la molecola di H 2, con 2 nuclei e due elettroni, si ha ( X 1,Y 1,Z 1,X 2,Y 2,Z 2 ;x 1,y 1,z 1,x 2,y 2,z 2 ) X 1,Y 1,Z 1 e X 2,Y 2,Z 2 coordinate dei nuclei 1 e 2 x 1,y 1,z 1,x 2,y 2,z 2 coordinate degli elettroni 1 e 2 2) Nell’equazione di Schrödinger compaiono anche le derivate rispetto alle coordinate nucleari
 La funzione d’onda dipende anche dalle coordinate nucleari oltre che da quelle elettroniche = (nuclei,elettroni) Ad esempio per la molecola di H 2, con 2 nuclei e due elettroni, si ha ( X 1,Y 1,Z 1,X 2,Y 2,Z 2 ;x 1,y 1,z 1,x 2,y 2,z 2 ) X 1,Y 1,Z 1 e X 2,Y 2,Z 2 coordinate dei nuclei 1 e 2 x 1,y 1,z 1,x 2,y 2,z 2 coordinate degli elettroni 1 e 2 2) Nell’equazione di Schrödinger compaiono anche le derivate rispetto alle coordinate nucleari")
22
Energia totale di un sistema di particelle in meccanica quantistica: molecole Questi due problemi sono risolti disaccoppiando il moto nucleare da quello elettronico in accordo con l’approssimazione di Born-Oppenheimer Le masse dei nuclei sono molto maggiori di quelle degli elettroni (1836 volte il protone) e quindi i nuclei sono molto più lenti. Di conseguenza gli elettroni possono aggiustarsi quasi istantaneamente ad ogni variazione delle posizioni dei nuclei. Da un punto di vista matematico ciò implica che la funzione d’onda può essere scritta nella forma (nuclei,elettroni) = (elettroni)· (nuclei) e si possono scrivere due equazioni di Schrödinger separate una per gli elettroni ed una per i nuclei
 = (elettroni)· (nuclei) e si possono scrivere due equazioni di Schrödinger separate una per gli elettroni ed una per i nuclei.")
23
Energia totale di un sistema di particelle in meccanica quantistica: molecole L’energia elettronica così ottenuta dipende indirettamente dalle coordinate nucleari e la funzione E el (nucleari) così ottenuta costituisce il potenziale nel quale si muovono i nuclei e che va inserito nell’equazione di Schrödinger per i nuclei Ĥ nucl (nuclei) = E nucl (nuclei) Poichè i nuclei sono particelle relativamente pesanti in pratica il moto nucleare può essere trattato con la meccanica classica cioè con le equazioni di Newton in cui il potenziale è proprio la funzione E el (nucleari) Quest’approssimazione è alla base del metodo della meccanica molecolare: i nuclei di una molecola sono considerati con la meccanica classica e la funzione di energia potenziale è trattata empiricamente come somma di semplici contributi (legami chimici, interazioni elettrostatiche,..) tra i vari atomi.
 così ottenuta costituisce il potenziale nel quale si muovono i nuclei e che va inserito nell’equazione di Schrödinger per i nuclei Ĥ nucl (nuclei) = E nucl (nuclei) Poichè i nuclei sono particelle relativamente pesanti in pratica il moto nucleare può essere trattato con la meccanica classica cioè con le equazioni di Newton in cui il potenziale è proprio la funzione E el (nucleari) Quest’approssimazione è alla base del metodo della meccanica molecolare: i nuclei di una molecola sono considerati con la meccanica classica e la funzione di energia potenziale è trattata empiricamente come somma di semplici contributi (legami chimici, interazioni elettrostatiche,..) tra i vari atomi.")
24
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare
 Potenziale elettrostatico Determinazione teorica della geometria molecolare")
25
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare La densità elettronica (r) può essere calcolata come somma dei quadrati degli orbitali molecolari occupati nel punto r.
 Potenziale elettrostatico Determinazione teorica della geometria molecolare La densità elettronica (r) può essere calcolata come somma dei quadrati degli orbitali molecolari occupati nel punto r.")
26
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare La reattività di una molecola dipende, nel caso di reazioni elettrofile, dalla localizzazione del valore più alto della probabilità di trovare l'elettrone nell'orbitale HOMO (highest occupied molecular orbital) e nel caso di reazioni nucleofile dalla localizzazione del valore più basso della probabilità nell'orbitale LUMO (lowest unoccupied molecular orbital). Gli orbitali di frontiera derivano da calcoli quantomeccanici e le rappresentazioni consistono in mappe di densità all'interno dell'orbitale. HOMO per la guanina

27
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare LUMO per la guanina La reattività di una molecola dipende, nel caso di reazioni elettrofile, dalla localizzazione del valore più alto della probabilità di trovare l'elettrone nell'orbitale HOMO (highest occupied molecular orbital) e nel caso di reazioni nucleofile dalla localizzazione del valore più basso della probabilità nell'orbitale LUMO (lowest unoccupied molecular orbital). Gli orbitali di frontiera derivano da calcoli quantomeccanici e le rappresentazioni consistono in mappe di densità all'interno dell'orbitale.

28
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare Il potenziale elettrostatico in un punto r è il lavoro fatto per portare una carica positiva unitaria dall'infinito a quel punto. Il potenziale elettrostatico molecolare (MEP) deriva quindi dall'interazione tra una distribuzione di carica (elettroni e nuclei) ed una carica puntiforme positiva unitaria, ed è quindi una misura della energia di interazione con un protone. Il potenziale elettrostatico per una data geometria può quindi essere calcolato in maniera rigorosa come la somma di un contributo dovuto alle cariche dei nuclei (N nuclei fissi posti in A) più un contributo dovuto agli elettroni. Poichè gli elettroni sono distribuiti in maniera continua nello spazio occorre sostituire l'integrale alla sommatoria ed introdurre la densità elettronica.
 deriva quindi dall interazione tra una distribuzione di carica (elettroni e nuclei) ed una carica puntiforme positiva unitaria, ed è quindi una misura della energia di interazione con un protone. Il potenziale elettrostatico per una data geometria può quindi essere calcolato in maniera rigorosa come la somma di un contributo dovuto alle cariche dei nuclei (N nuclei fissi posti in A) più un contributo dovuto agli elettroni. Poichè gli elettroni sono distribuiti in maniera continua nello spazio occorre sostituire l integrale alla sommatoria ed introdurre la densità elettronica..")
29
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare Per i sistemi di interesse biologico è impensabile calcolare il potenziale dalla funzione d'onda quantomeccanica per il numero notevole di atomi di questi sistemi, che rendono il calcolo impossibile. Si fa perciò ricorso a metodi approssimati, derivati e controllati nel corso degli anni. Il potenziale può essere derivato con buona approssimazione in modo additivo da contributi rappresentabili mediante opportuni sistemi di cariche puntiformi poste nelle posizioni occupate dagli atomi. Il valore delle cariche puntiformi può in un certo senso essere considerato dovuto al bilanciamento tra carica nucleare ed elettronica, attribuibili a quell'atomo, I valori numerici delle cariche puntiformi possono essere ottenuti da calcoli quantomeccanici o da metodi approssimati

30
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare Il MEP è una proprietà locale (dipende dal punto in cui viene valutata) calcolabile in un reticolo tridimensionale di punti intorno alla molecola. Il MEP può essere codificato, mediante almeno tre colori, anche sulla superficie molecolare (superficie di van der Waals o sulla superficie accessibile al solvente) come rappresentato in figura.
 come rappresentato in figura..")
31
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare Potenziale elettrostatico mappato sulla superficie di isodensità elettronica per la formammide. Il colore rosso indica un potenziale elettrostatico negativo e il blu un potenziale elettrostatico positivo.

32
Proprietà molecolari Densità elettronica Orbitali (HOMO, LUMO) Potenziale elettrostatico Determinazione teorica della geometria molecolare In figura (a) è riportato il MEP sulla superficie molecolare di un pirrolo dove i valori più negativi (-38 Kcal/mol) corrispondono alla zona rossa. Nella figura (b) è riportata la superficie di isoenergia (a –35 Kcal/mol) del MEP ancora sul pirrolo.
 è riportata la superficie di isoenergia (a –35 Kcal/mol) del MEP ancora sul pirrolo..")
Presentazioni simili

 Neutroni (n°) Elettroni (e) Gli atomi contengono diversi tipi di particelle subatomiche.>")














 per riprodurre e “mimare” il comportamento di molecole e di sistemi molecolari. C'è.>")


 r rmrm εmεm r=σ Regione attrattiva Regione repulsiva V(r m )=-ε, F attr =F rep V(σ)=0, V attr =V rep.>")