Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
A genetic algorithm for the ligand-protein docking problem
Research article Genetics and Molecular Biology (2004)
")
2
DOCKING E STRUCTURE-BASED RATIONAL DRUG DISCOVERY AND DESIGN

3
IL DOCKING strutture 3D conosciute
predire se e come interagiranno per formare un complesso molecolare funzionale Metodi energetici Metodi geometrici Proteina: in complesso e isolata; molteplici stati conformazionali;

4
RIGID AND FLEXIBLE DOCKING
DNA-Protein docking Protein-Protein docking Ligand-Protein Docking:

5
FORZE CHIMICO-FISICHE DOCKING SCORES
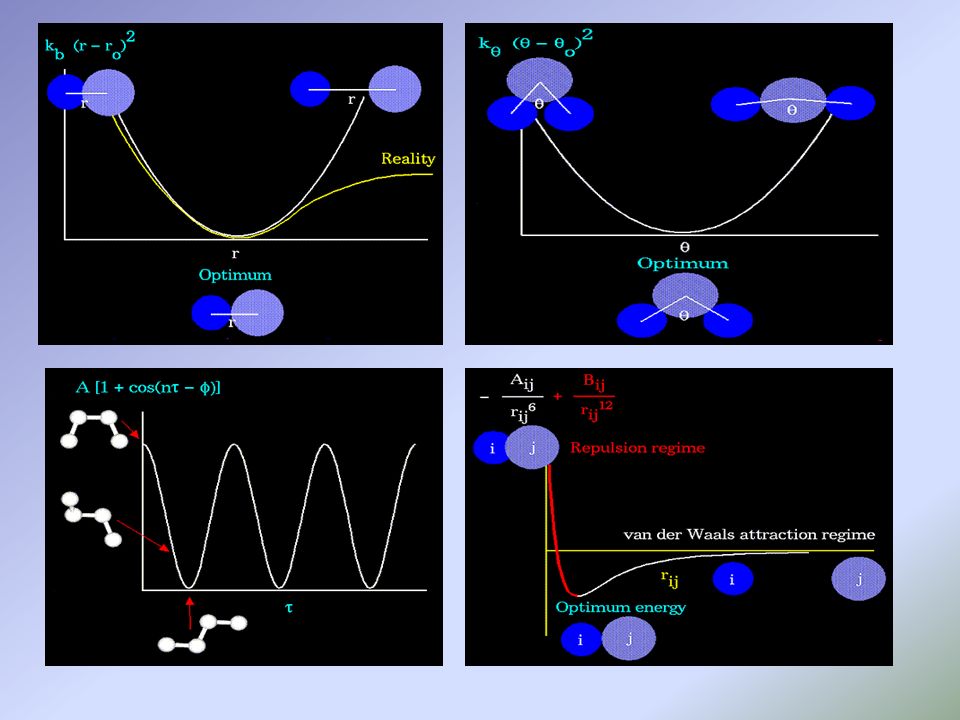
stabilità delle forze plausibilità della soluzione MISURA LA QUALITA’ DELLA SOLUZIONE PROPOSTA Interazioni elettrostatiche Forze di Van der Walls E = A/(r6) - B/(r12) Legami a idrogeno
 - B/(r12) Legami a idrogeno")
6
DRUG DESIGN KEY-LOCK MODEL
Attacco Penetrazione Spoliazione Trascrizione Replicazione Traduzione dei messaggeri Assemblaggio Egresso

7
DRUG DESIGN KEY-LOCK MODEL
HIV VIRUS TARGET-LOCK Trascrittasi inversa Proteasi virale Current DRUG-KEY AZT Inibitori delle proteasi

8
Attività della proteasi
HIV-protease ( x-ray, 1989) Attività della proteasi Elaborazione proteolitica delle proteine strutturali ed enzimatiche per l’assemblaggio del virione
 Attività della proteasi. Elaborazione proteolitica delle proteine strutturali ed enzimatiche per l’assemblaggio del virione.")
9
MATURAZIONE DELLA PARTICELLA VIRALE:HIV
Virione maturo Virione immaturo PROTEASI VIRALE Gli INIBITORI DELLE PROTEASI impediscono la maturazione della particella virale

10
DOCKING SCREEN INPUT PROGRAM Search algorithm Scoring function
Struttura 3D della proteina di interesse (target) Database di potenziali ligandi (drug) PROGRAM Search algorithm Scoring function
 Database di potenziali ligandi (drug) PROGRAM. Search algorithm. Scoring function.")
11
THE SEARCH SPACE TUTTI i possibili orientamenti e le possibili conformazioni della coppia di molecole Simulazioni di propongono istantanee dinamica molecolare energeticamente favorevoli Combinazioni di diverse simulano flessibilità strutture dello stesso target del recettore ALGORITMI GENETICI interazioni sempre più favorevoli

12
molecular mechanics force field (E)
THE SCORING FUNCTION Input : istantanea della coppia Output: valore di probabilità che l’input sia un’interazione favorevole molecular mechanics force field (E) Potenziale che descrive le caratteristiche energetiche delle molecole in relazione a ciò che le circonda
 Potenziale che descrive le caratteristiche energetiche delle molecole in relazione a ciò che le circonda.")
13
E = Estr + Ebend + Etors + Evdw + Eel + Ecross

15
GROMOS Class I Methods: Class II Methods:
Higher order terms and cross terms. Higher accuracy, used for small or medium sized molecules. Parameterized from experimental data. Class II Methods: For very large molecules,only quadratic Taylor expansions and neglecting cross terms. GROMOS

16
GROMOS Pacchetto di simulazione di dinamica molecolare per studiare sistemi biologici In soluzione o cristallizzati Minimizzazione di energia Analisi delle conformazioni ottenute sperimentalmente o tramite simulazioni Applicazioni: Predizione di conformazioni Predizione di variazioni energetiche e conformazionali al variare di aa o bp Derivazioni di strutture 3D Modellizzazione dinamica di complessi tramite ricerca nello spazio delle configurazioni Predizione di proprietà in condizioni estreme di T e P

17
IL PROBLEMA: come trattare la flessibilità in entrambe le molecole!
Ligando e recettore come molecole rigide Flessibilità del ligando THE CHALLENGE Docking di molecole grandi e altamente flessibili Flessibilità del recettore

18
LA METAFORA EVOLUZIONE PROBLEM SOLVING Ambiente Problema da risolvere
Individuo Soluzione candidata Addattamento Qualità della soluzione

19
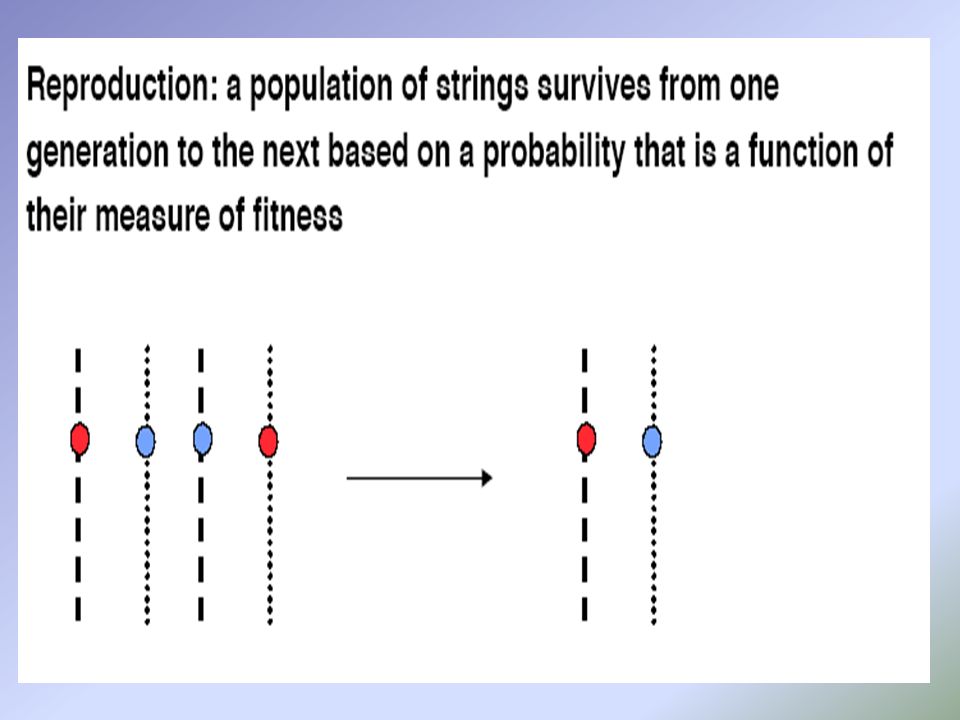
GLI ALGORITMI GENETICI
Un semplice GA consiste di tre operazioni: selezione,operazioni genetiche e sostituzione.

23
STEADY-STATE REPLACEMENT
Solo pochi cromosomi vengono sostituiti dalla prole, i peggiori, gli altri continuano a far parte della popolazione A ogni generazione è applicato un solo operatore genetico per volta e viene sostituito un solo individuo

24
IN PARTICOLARE… Grid – based methodology in the rigid and flexible ligand-docking cases Rigid receptor structure Highly flexible ligands, more than 10 conformational degrees of freedom 5 HIV-1 protease-ligand complexes ( known 3D )
")
25
Implemented SSGA Ogni cromosoma ha 3 geni per ligand translation (coordinate atomiche X Y Z ) 4 geni per ligand orientation Altri per ligand conformation (angoli diedri del ligando) Ligand-protein energy function used is the GROMOS96 classical force field
 Ligand-protein energy function used is the GROMOS96 classical force field.")
26
The grid-based methodology
Il sito attivo della proteina è rappresentato all’interno di una griglia rettangolare 3D Ogni celletta ha un punteggio in relazione alla distanza dagli atomi del sito attivo Per ogni conformazione del ligando proposta viene calcolato il punteggio La griglia utilizzata è la media tra tutte le griglie corrispondenti a ogni possibile conformazione del recettore

27
Implemented SSGA La popolazione iniziale di individui è generata casualmente all’interno della griglia con valori compresi tra la minima e la massima dimensione delle griglie Gli individui per le generazioni successive sono poi generati attraverso perturbazioni casuali delle coordinate dei geni di traslazione Per i geni che corrispondono ad angoli sono generati valori casuali compresi tra 0° e 360° Per i geni di orientamento sono generati valori compresi tra -1 e +1

28
Implemented SSGA I nuovi individui sono poi selezionati e mutati o ricombinati Un nuovo individuo viene inserito nella popolazione se la sua fitness è migliore di quella peggiore della popolazione corrente L’algoritmo procede finchè non è stato raggiunto il massimo numero di valutazioni energetiche

29
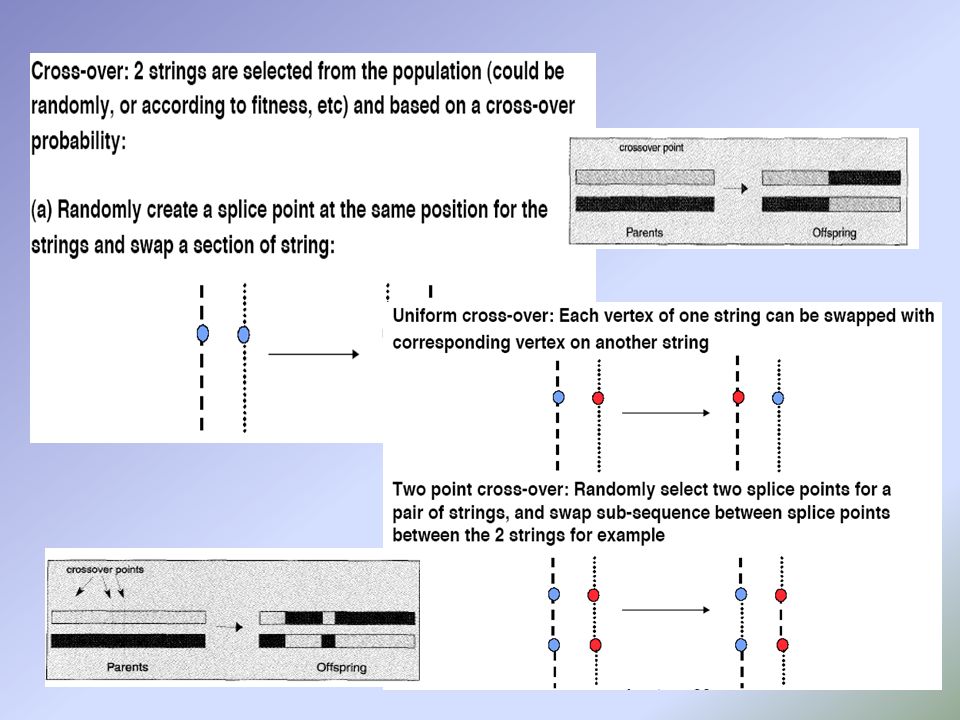
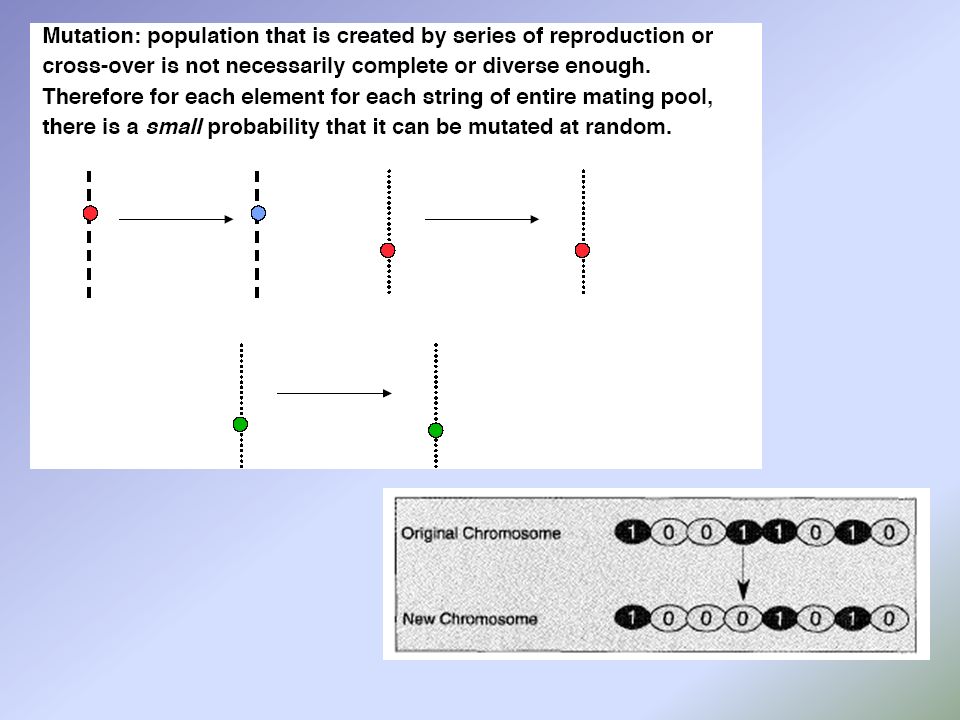
Two – point crossing over e Non - uniform mutation
a e b sono i limiti superiori e inferiori della variabile c τ è scelto casualmente tra 0 e 1 b è settato a 5

30
I ligandi testati hanno da 12 a 20 angoli conformazionali (diedri) la griglia è stata centrata nel sito attivo della proteina Le frecce curve indicano gli angoli diedri Le frecce dritte indicano gli atomi di riferimento

31
A ligand in the HIV-1 protease active site
radice quadratica media della deviazione tra gli atomi del modello e della proteina dopo che le due strutture sono state sovrapposte nel modo migliore possibile A ligand in the HIV-1 protease active site Il successo dell’algoritmo è stato misurato attraverso il RMSD (root mean square deviation) tra la conformazione cristallografica (PDB file) e quella trovata dall’algoritmo RMSD < 2Ǻ = DOCKED … risultato molto buono RMSD < 3Ǻ = partially DOCKED La % di successo è il numero di conformazioni trovate con RMSD<2Ǻ in 10 tentativi
 tra la conformazione cristallografica (PDB file) e quella trovata dall’algoritmo. RMSD < 2Ǻ = DOCKED … risultato molto buono. RMSD < 3Ǻ = partially DOCKED. La % di successo è il numero di conformazioni trovate con RMSD<2Ǻ in 10 tentativi.")
32
RIGID DOCKING TESTS Angoli diedri dei ligandi fissi in posizione “cristallografica” Movimenti di traslazione e orientamento i cromosomi hanno solo i geni di traslazione e orientamento e gli ultimi due termini della funzione energetica non sono considerati 500 individui 200,000 valutazioni energetiche p = 0,3 per crossing-over p= 0,7 per mutazione

33
ENERGIA MEDIA (Kcal/mol)
LIGANDI DMP323 NELFINAVIR RITONAVIR INDINAVIR SAQUINAVIR RMSD ME DIA (Ǻ) 0.046 0.068 0.099 0.053 0.077 ENERGIA MEDIA (Kcal/mol) -58 -82 -100 -87 % DI SUCCESSO 100 Lo SSGA è in grado di trovare le conformazioni corrispondenti alle strutture cristallografiche per tutti i ligandi testati
 ENERGIA MEDIA (Kcal/mol) % DI SUCCESSO Lo SSGA è in grado di trovare le conformazioni corrispondenti alle strutture cristallografiche per tutti i ligandi testati.")
34
FLEXIBLE DOCKING TESTS
Tutti i termini energetici sono stati considerati 1,000 individui 1,000,000 di valutazioni energetiche p = 0,3 per crossing-over p= 0,7 per mutazione

35
DMP323 flexible docking results
ANGOLI DIEDRI CONSIDERATI 10 14 RMSD MEDIO (Ǻ) 0,373 0,596 ENERGIA MEDIA (Kcal/mol) -32 -33 % DI SUCCESSO 100
 0,373. 0,596. ENERGIA MEDIA (Kcal/mol) % DI SUCCESSO")
36
flexible docking results
LIGANDI DMP323 NELFINAVIR RITONAVIR INDINAVIR SAQUINAVIR ANGOLI DIEDRI CONSIDERATI 14 12 20 15 RMSD ME DIA (Ǻ) 0.596 4.185 4.237 5.755 3.585 ENERGIA MEDIA (Kcal/mol) -32 -26 -41 36 -19 % DI SUCCESSO per rmsd < di 2 e 3 Ǻ 100 30 50 10 60
 ENERGIA MEDIA (Kcal/mol) % DI SUCCESSO per rmsd < di 2 e 3 Ǻ")
37
flexible docking results fissando alcuni angoli diedri più interni
LIGANDI DMP323 NELFINAVIR RITONAVIR INDINAVIR SAQUINAVIR ANGOLI DIEDRI CONSIDERATI 14 10 17 12 13 RMSD ME DIA (Ǻ) 0.596 1.449 3.733 3.118 3.106 ENERGIA MEDIA (Kcal/mol) -32 -53 -70 -17 -26 % DI SUCCESSO per rmsd < di 2 e 3 Ǻ 100 90 30 60 20 50
 ENERGIA MEDIA (Kcal/mol) % DI SUCCESSO per rmsd < di 2 e 3 Ǻ")
38
CONCLUSIONI La performance migliore di DMP323 rispetto agli altri ligandi può dipendere dalla minore dipendenza tra i suoi angoli diedri e dal fatto che la sua esatta conformazione è al centro del sito attivo della proteina Gli altri ligandi hanno una geometria più “aperta” e quindi maggior dipendenda tra gli angoli diedri, soprattutto quelli più interni Questo sembra essere dovuto al fatto che variazioni negli angoli interni provocano variazioni maggiori nell’intera molecola

39
CONCLUSIONI I risultati ottenuti mostrano la difficoltà di trattare con ligandi altamente flessibili, cioè con molti gradi di libertà conformazionali Tutti i programmi di docking attuali mostrano una performance che diminuisce con l’aumentare del numero di gradi di libertà conformazionale considerati SSGA: buono per docking rigido (5 minuti) da migliorare per il docking flessibile Servono nuovi operatori che prendano in considerazione i cambiamenti degli angoli diedri più interni
 da migliorare per il docking flessibile. Servono nuovi operatori che prendano in considerazione i cambiamenti degli angoli diedri più interni.")
Presentazioni simili