Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni Genomiche: (±Ch). Sono dovute a non disgiunzioni meiotiche (es. Trisomia 21). Genomiche: (±Ch). Sono dovute a non disgiunzioni meiotiche (es. Trisomia 21). Cromosomiche: Traslocazioni, amplificazioni, macrodelezioni. Cromosomiche: Traslocazioni, amplificazioni, macrodelezioni. Geniche: microdelezioni, sostituzioni, inserzioni di uno o pochissimi nucleotidi (puntiformi). Associate a malattie ereditarie Mendeliane. Geniche: microdelezioni, sostituzioni, inserzioni di uno o pochissimi nucleotidi (puntiformi). Associate a malattie ereditarie Mendeliane.
. Genomiche: (±Ch). Sono dovute a non disgiunzioni meiotiche (es. Trisomia 21). Cromosomiche: Traslocazioni, amplificazioni, macrodelezioni. Cromosomiche: Traslocazioni, amplificazioni, macrodelezioni. Geniche: microdelezioni, sostituzioni, inserzioni di uno o pochissimi nucleotidi (puntiformi). Associate a malattie ereditarie Mendeliane. Geniche: microdelezioni, sostituzioni, inserzioni di uno o pochissimi nucleotidi (puntiformi). Associate a malattie ereditarie Mendeliane..")
2
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE GENETICHE Malattie genomiche e cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie genomiche e cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale: si trasmettono solo in linea femminile. Malattie legate ad eredità mitocondriale: si trasmettono solo in linea femminile.
. Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale: si trasmettono solo in linea femminile. Malattie legate ad eredità mitocondriale: si trasmettono solo in linea femminile..")
3
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Malattie genomiche UOMO 2n=46 ( 46,XY- 46,XX) UOMO 2n=46 ( 46,XY- 46,XX) Lyon: I cromosomi X oltre il primo, vengono selezionati a caso ed inattivati per metilazione del DNA (corpi di Barr). Nelle femmine il 50% di X deriva dalla madre ed il 50% dal padre. Lyon: I cromosomi X oltre il primo, vengono selezionati a caso ed inattivati per metilazione del DNA (corpi di Barr). Nelle femmine il 50% di X deriva dalla madre ed il 50% dal padre. I cromosomi Y si vedono all U.V. con chinacrina. I cromosomi Y si vedono all U.V. con chinacrina.
. Nelle femmine il 50% di X deriva dalla madre ed il 50% dal padre. I cromosomi Y si vedono all U.V. con chinacrina. I cromosomi Y si vedono all U.V. con chinacrina..")
4
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

5
CARIOTIPO Euploide: numero normale di Ch. Euploide: numero normale di Ch. Diploide:2n Ch nelle cellule somatiche (46) Diploide:2n Ch nelle cellule somatiche (46) Aploide: n Ch nelle cellule germinali (23) Aploide: n Ch nelle cellule germinali (23) Poliploide: aumento numerico del corredo di Ch (3n,4n ecc.). La poliploidia nel feto causa aborto, avviene nella rigenerazione e nei tumori. Poliploide: aumento numerico del corredo di Ch (3n,4n ecc.). La poliploidia nel feto causa aborto, avviene nella rigenerazione e nei tumori. Aneuploide: Ch multipli imperfetti di n (23) (Down 47, Turner 45) Aneuploide: Ch multipli imperfetti di n (23) (Down 47, Turner 45)
 Diploide:2n Ch nelle cellule somatiche (46) Aploide: n Ch nelle cellule germinali (23) Aploide: n Ch nelle cellule germinali (23) Poliploide: aumento numerico del corredo di Ch (3n,4n ecc.). La poliploidia nel feto causa aborto, avviene nella rigenerazione e nei tumori. Poliploide: aumento numerico del corredo di Ch (3n,4n ecc.). La poliploidia nel feto causa aborto, avviene nella rigenerazione e nei tumori. Aneuploide: Ch multipli imperfetti di n (23) (Down 47, Turner 45) Aneuploide: Ch multipli imperfetti di n (23) (Down 47, Turner 45).")
6
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 ANEUPLOIDIE Polisomia: più di due Ch omologhi (Down 47XY+21, 47XX+21) Polisomia: più di due Ch omologhi (Down 47XY+21, 47XX+21) Monosomia: mancanza di uno dei due Ch omologhi (Turner 45,X) Monosomia: mancanza di uno dei due Ch omologhi (Turner 45,X) Non disgiunzione meiotica: gameti con 24Ch + gameti con 22Ch. Fecondati da gameti normali (23 Ch) danno trisomie e monosomie. Non disgiunzione meiotica: gameti con 24Ch + gameti con 22Ch. Fecondati da gameti normali (23 Ch) danno trisomie e monosomie. Non disgiunzione mitotica: Mosaicismo con popolazioni geneticamente diverse. Non disgiunzione mitotica: Mosaicismo con popolazioni geneticamente diverse.
 danno trisomie e monosomie. Non disgiunzione meiotica: gameti con 24Ch + gameti con 22Ch. Fecondati da gameti normali (23 Ch) danno trisomie e monosomie. Non disgiunzione mitotica: Mosaicismo con popolazioni geneticamente diverse. Non disgiunzione mitotica: Mosaicismo con popolazioni geneticamente diverse..")
7
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

8
Non disgiunzione: cause Radiazioni ionizzanti Radiazioni ionizzanti Malattie virali (epatite, morbillo) Malattie virali (epatite, morbillo) Tiroidite autoimmune Tiroidite autoimmune ETA MATERNA : ETA MATERNA : DOWN DOWN 1/192520 aa 1/192520 aa 1/88530aa 1/88530aa 1/38535aa 1/38535aa 1/1149aa 1/1149aa
 Malattie virali (epatite, morbillo) Tiroidite autoimmune Tiroidite autoimmune ETA MATERNA : ETA MATERNA : DOWN DOWN 1/ aa 1/ aa 1/88530aa 1/88530aa 1/38535aa 1/38535aa 1/1149aa 1/1149aa")
9
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 TRISOMIE AUTOSOMICHE (I) Trisomia 21(Sindrome di Down) Trisomia 21(Sindrome di Down) E relativamente ben tollerata E relativamente ben tollerata 2/3 aborto spontaneo o morte in utero 2/3 aborto spontaneo o morte in utero Ridotte aspettative di vita Ridotte aspettative di vita
 Trisomia 21(Sindrome di Down) Trisomia 21(Sindrome di Down) E relativamente ben tollerata E relativamente ben tollerata 2/3 aborto spontaneo o morte in utero 2/3 aborto spontaneo o morte in utero Ridotte aspettative di vita Ridotte aspettative di vita")
10
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 TRISOMIE AUTOSOMICHE (II) Difetti cardiaci (settali, atriali, ventricolari) Difetti cardiaci (settali, atriali, ventricolari) Difetti gastrointestinali (fistole tracheo- esofagee, atresia duodenale) Difetti gastrointestinali (fistole tracheo- esofagee, atresia duodenale) Ematologici (>LMA, >LLA) Ematologici (>LMA, >LLA) Morbo di Alzheimer Morbo di Alzheimer Risposte immunitarie alterate (> suscettibilità alle infezioni e alle tiroiditi autoimmuni) Risposte immunitarie alterate (> suscettibilità alle infezioni e alle tiroiditi autoimmuni) sterili; fertili sterili; fertili
 Difetti cardiaci (settali, atriali, ventricolari) Difetti cardiaci (settali, atriali, ventricolari) Difetti gastrointestinali (fistole tracheo- esofagee, atresia duodenale) Difetti gastrointestinali (fistole tracheo- esofagee, atresia duodenale) Ematologici (>LMA, >LLA) Ematologici (>LMA, >LLA) Morbo di Alzheimer Morbo di Alzheimer Risposte immunitarie alterate (> suscettibilità alle infezioni e alle tiroiditi autoimmuni) Risposte immunitarie alterate (> suscettibilità alle infezioni e alle tiroiditi autoimmuni) sterili; fertili sterili; fertili")
11
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 TRISOMIE AUTOSOMICHE (III) FACIES MONGOLOIDES FACIES MONGOLOIDES Profilo facciale appiattito Profilo facciale appiattito Pieghe epicantiche prominenti Pieghe epicantiche prominenti Pelle del collo ridondante Pelle del collo ridondante Macroglossia Macroglossia Ernia ombelicale Ernia ombelicale Ipotonia muscolare Ipotonia muscolare Spazio fra il I e il II dito del piede Spazio fra il I e il II dito del piede
 FACIES MONGOLOIDES FACIES MONGOLOIDES Profilo facciale appiattito Profilo facciale appiattito Pieghe epicantiche prominenti Pieghe epicantiche prominenti Pelle del collo ridondante Pelle del collo ridondante Macroglossia Macroglossia Ernia ombelicale Ernia ombelicale Ipotonia muscolare Ipotonia muscolare Spazio fra il I e il II dito del piede Spazio fra il I e il II dito del piede")
12
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

13
TRISOMIE AUTOSOMICHE(VI) Trisomia 18 (Sindrome di Edward) (Incidenza 1:8000) Trisomia 18 (Sindrome di Edward) (Incidenza 1:8000) Simile alla sinrome di Down Simile alla sinrome di Down 90% di mortalità nel primo mese di vita 90% di mortalità nel primo mese di vita Alterato sviluppo cerebrale Alterato sviluppo cerebrale Difetti del setto ventricolare Difetti del setto ventricolare
 Trisomia 18 (Sindrome di Edward) (Incidenza 1:8000) Trisomia 18 (Sindrome di Edward) (Incidenza 1:8000) Simile alla sinrome di Down Simile alla sinrome di Down 90% di mortalità nel primo mese di vita 90% di mortalità nel primo mese di vita Alterato sviluppo cerebrale Alterato sviluppo cerebrale Difetti del setto ventricolare Difetti del setto ventricolare")
14
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 TRISOMIE AUTOSOMICHE (V) Trisomia 13 (Sindrome di Patau) Trisomia 13 (Sindrome di Patau) (Incidenza 1:15000) (Incidenza 1:15000) Simile alla sindrome di Down Simile alla sindrome di Down 80% di mortalità nel primo anno di vita 80% di mortalità nel primo anno di vita Gravi malformazioni facciali Gravi malformazioni facciali Grave ritardo mentale Grave ritardo mentale Difetti del setto ventricolare Difetti del setto ventricolare Labbro leporino e palatoschisi Labbro leporino e palatoschisi
 Trisomia 13 (Sindrome di Patau) Trisomia 13 (Sindrome di Patau) (Incidenza 1:15000) (Incidenza 1:15000) Simile alla sindrome di Down Simile alla sindrome di Down 80% di mortalità nel primo anno di vita 80% di mortalità nel primo anno di vita Gravi malformazioni facciali Gravi malformazioni facciali Grave ritardo mentale Grave ritardo mentale Difetti del setto ventricolare Difetti del setto ventricolare Labbro leporino e palatoschisi Labbro leporino e palatoschisi")
15
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

16
Trisomia del cromosoma 13

17
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 ANEUPLOIDIE DEI Ch SESSUALI (I) Sono meglio tollerate delle autosomiche Relativamente frequenti Solo 45,Y0 é disvitale
 Sono meglio tollerate delle autosomiche Relativamente frequenti Solo 45,Y0 é disvitale")
18
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 ANEUPLOIDIE DEI Ch SESSUALI (II) Sindrome di Turner (incidenza 1: 7000) Sindrome di Turner (incidenza 1: 7000) 60%45,X0 40% mosaici (X0/46,XY) e isocromosomia X 60%45,X0 40% mosaici (X0/46,XY) e isocromosomia X Neonati fenotipicamente Neonati fenotipicamente Dilatazione dei vasi linfatici Dilatazione dei vasi linfatici Bassa statura, alterazioni di orecchie, bocca e collo Bassa statura, alterazioni di orecchie, bocca e collo Amenorrea primaria Amenorrea primaria Q.I. Pressocchè normale Q.I. Pressocchè normale

19
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 bassa statura, infantilismo sessuale e sterilità. malformazioni cardiache, renali, aspetti somatici peculiari Sindrome di Turner

20
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 ANEUPLOIDIE DEI Ch SESSUALI (III) Sindrome di Klinefelter (incidenza 1:850) 47,XXY con corpo di Barr. Sindrome di Klinefelter (incidenza 1:850) 47,XXY con corpo di Barr. Neonati fenotipicamente Neonati fenotipicamente Proporzioni eunucoidi, abitus longilineo, ipogonadismo con testicoli atrofici Proporzioni eunucoidi, abitus longilineo, ipogonadismo con testicoli atrofici Ginecomastia (25%) Ginecomastia (25%) Qualche problema di apprendimento Qualche problema di apprendimento
 47,XXY con corpo di Barr. Neonati fenotipicamente Neonati fenotipicamente Proporzioni eunucoidi, abitus longilineo, ipogonadismo con testicoli atrofici Proporzioni eunucoidi, abitus longilineo, ipogonadismo con testicoli atrofici Ginecomastia (25%) Ginecomastia (25%) Qualche problema di apprendimento Qualche problema di apprendimento.")
21
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Sindrome di Klinefelter Sindrome di Klinefelter frequenza alla nascita di 1:1000 Ipogonadismo bassi livelli di testosterone, mancata produzione di spermatozoi (azoospermia) e quindi sterilità; sproporzione tra lunghezza degli arti e lunghezza del tronco Altezza superiore alla media
 e quindi sterilità; sproporzione tra lunghezza degli arti e lunghezza del tronco Altezza superiore alla media")
22
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 ALTRE ANEUPLOIDIE DEI Ch SESSUALI (VI) 47,XYY: alta staura, aggressività, basso QI 47,XYY: alta staura, aggressività, basso QI 47,XXX: fenotipicamente silente 47,XXX: fenotipicamente silente 48,XXXX: ritardo mentale ± grave 48,XXXX: ritardo mentale ± grave 49,XXXXX: grave ritardo mentale, anomalie somatiche 49,XXXXX: grave ritardo mentale, anomalie somatiche 48, XXYY, 48,XXXY, 49,XXXYY, 49,XXXXY sono varianti di Klinefelter 48, XXYY, 48,XXXY, 49,XXXYY, 49,XXXXY sono varianti di Klinefelter
 47,XYY: alta staura, aggressività, basso QI 47,XYY: alta staura, aggressività, basso QI 47,XXX: fenotipicamente silente 47,XXX: fenotipicamente silente 48,XXXX: ritardo mentale ± grave 48,XXXX: ritardo mentale ± grave 49,XXXXX: grave ritardo mentale, anomalie somatiche 49,XXXXX: grave ritardo mentale, anomalie somatiche 48, XXYY, 48,XXXY, 49,XXXYY, 49,XXXXY sono varianti di Klinefelter 48, XXYY, 48,XXXY, 49,XXXYY, 49,XXXXY sono varianti di Klinefelter")
23
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 ALTRE MALATTIE CROMOSOMICHE Sindrome del Cri du chat: Del. Braccio corto del Ch 5 (ritardo mentale) Sindrome del Cri du chat: Del. Braccio corto del Ch 5 (ritardo mentale) Sindrome di Prader-Willi: microdelezione del Ch.15 derivato dal padre (ritardo mentale, ipotonicità, obesità) Sindrome di Prader-Willi: microdelezione del Ch.15 derivato dal padre (ritardo mentale, ipotonicità, obesità) Sindrome di Angelman: microdelezione del Ch.15 derivato dalla madre (ritardo mentale, convulsioni, scoppi di risa) Sindrome di Angelman: microdelezione del Ch.15 derivato dalla madre (ritardo mentale, convulsioni, scoppi di risa)
 Sindrome del Cri du chat: Del. Braccio corto del Ch 5 (ritardo mentale) Sindrome di Prader-Willi: microdelezione del Ch.15 derivato dal padre (ritardo mentale, ipotonicità, obesità) Sindrome di Prader-Willi: microdelezione del Ch.15 derivato dal padre (ritardo mentale, ipotonicità, obesità) Sindrome di Angelman: microdelezione del Ch.15 derivato dalla madre (ritardo mentale, convulsioni, scoppi di risa) Sindrome di Angelman: microdelezione del Ch.15 derivato dalla madre (ritardo mentale, convulsioni, scoppi di risa).")
24
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MUTAZIONI CROMOSOMICHE E GENICHE

25
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazione: alterazione stabile ed ereditabile del DNA Non sempre é un evento negativo. Non sempre é un evento negativo. Può essere spontanea (molto rara) o indotta (agenti chimici, fisici o virali). Può essere spontanea (molto rara) o indotta (agenti chimici, fisici o virali). Se coinvolge le cellule germinali viene trasmessa alla progenie (Malattie ereditarie) Se coinvolge le cellule germinali viene trasmessa alla progenie (Malattie ereditarie) Se coinvolge cellule somatiche può essere implicata nella tumorigenesi Se coinvolge cellule somatiche può essere implicata nella tumorigenesi
 o indotta (agenti chimici, fisici o virali). Può essere spontanea (molto rara) o indotta (agenti chimici, fisici o virali). Se coinvolge le cellule germinali viene trasmessa alla progenie (Malattie ereditarie) Se coinvolge le cellule germinali viene trasmessa alla progenie (Malattie ereditarie) Se coinvolge cellule somatiche può essere implicata nella tumorigenesi Se coinvolge cellule somatiche può essere implicata nella tumorigenesi.")
26
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni cromosomiche:TRASLOCAZIONE Mutazione cromosomica che modifica la posizione di un segmento cromosomico e delle sequenze geniche in esso contenute. Mutazione cromosomica che modifica la posizione di un segmento cromosomico e delle sequenze geniche in esso contenute. Intracromosomica o fra Ch non omologhi Intracromosomica o fra Ch non omologhi Semplice: trasporto di un segmento terminale a un Ch non omologo Semplice: trasporto di un segmento terminale a un Ch non omologo Reciproca: due break point in due Ch con scambio di materiale Reciproca: due break point in due Ch con scambio di materiale Bilanciata: senza perdita di materiale genetico (funzionante) Bilanciata: senza perdita di materiale genetico (funzionante)
 Bilanciata: senza perdita di materiale genetico (funzionante).")
27
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

28
Mutazioni cromosomiche:TRASLOCAZIONE ROBERTSONIANA Reciproca, bilanciata fra due Ch acrocentrici (13,14,21). Si perde solo materiale non tradotto e si ottiene un Ch grande dalla fusione dei due bracci lunghi (funzionanti). Soggetto normale con 45 Ch. Reciproca, bilanciata fra due Ch acrocentrici (13,14,21). Si perde solo materiale non tradotto e si ottiene un Ch grande dalla fusione dei due bracci lunghi (funzionanti). Soggetto normale con 45 Ch. Rischio di Down nella progenie Rischio di Down nella progenie
. Soggetto normale con 45 Ch. Reciproca, bilanciata fra due Ch acrocentrici (13,14,21). Si perde solo materiale non tradotto e si ottiene un Ch grande dalla fusione dei due bracci lunghi (funzionanti). Soggetto normale con 45 Ch. Rischio di Down nella progenie Rischio di Down nella progenie.")
29
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni cromosomiche:DELEZIONE Mutazione cromosomica con perdita di un tratto di Ch. Apicale o Intercalare. Mutazione cromosomica con perdita di un tratto di Ch. Apicale o Intercalare. Il danno dipende dal materiale perso. Il danno dipende dal materiale perso. Ch ad anello: Del di frammenti apicali del braccio lungo e del braccio corto. Ch ad anello: Del di frammenti apicali del braccio lungo e del braccio corto. Isocromosoma: Del di un braccio lungo o corto + duplicazione del residuo. Isocromosoma: Del di un braccio lungo o corto + duplicazione del residuo.

30
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni cromosomiche:DUPLICAZIONE Mutazione cromosomica con raddoppio di un tratto di Ch. Mutazione cromosomica con raddoppio di un tratto di Ch. Importante nello sviluppo di famiglie geniche (es. Globina). Importante nello sviluppo di famiglie geniche (es. Globina). Tratti duplicati vicini (tandem) o distanti. Tratti duplicati vicini (tandem) o distanti. Conseguenze legate all eccesso di materiale genico e alla difficoltà di appaiamento degli omologhi (es. Neuropatia demielinizzante da duplicazione del gene PM22) Conseguenze legate all eccesso di materiale genico e alla difficoltà di appaiamento degli omologhi (es. Neuropatia demielinizzante da duplicazione del gene PM22)
. Importante nello sviluppo di famiglie geniche (es. Globina). Tratti duplicati vicini (tandem) o distanti. Tratti duplicati vicini (tandem) o distanti. Conseguenze legate all eccesso di materiale genico e alla difficoltà di appaiamento degli omologhi (es. Neuropatia demielinizzante da duplicazione del gene PM22) Conseguenze legate all eccesso di materiale genico e alla difficoltà di appaiamento degli omologhi (es. Neuropatia demielinizzante da duplicazione del gene PM22).")
31
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni cromosomiche:INVERSIONE Pericentrica o paracentrica Pericentrica o paracentrica Ha conseguenze durante i crossing – over che avvengono tra due cromatidi omologhi non fratelli. Ha conseguenze durante i crossing – over che avvengono tra due cromatidi omologhi non fratelli.

32
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Meccanismi patogenetici delle mutazioni geniche Alterazione strutturale di una proteina con formazione di un prodotto anomalo. Alterazione strutturale di una proteina con formazione di un prodotto anomalo. -Carenza del prodotto normale; -Prodotto alterato che interferisce con quello normale; -prodotto alterato con funzione diversa. Alterazione quantitativa (riduzione o abolizione di una proteina WT o overspressione di essa), per mutazioni che riguardano le sequenze di splicing o dei promotori. Alterazione quantitativa (riduzione o abolizione di una proteina WT o overspressione di essa), per mutazioni che riguardano le sequenze di splicing o dei promotori.
, per mutazioni che riguardano le sequenze di splicing o dei promotori. Alterazione quantitativa (riduzione o abolizione di una proteina WT o overspressione di essa), per mutazioni che riguardano le sequenze di splicing o dei promotori..")
33
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Effetti delle mutazioni geniche Eterogeneità genetica allelica: mutazioni#, effetti= sulla stessa pt. (es.mutazioni della G6P deH); Eterogeneità genetica allelica: mutazioni#, effetti= sulla stessa pt. (es.mutazioni della G6P deH); Eterogeneità genetica non allelica: mutazioni di pt # coinvolte nello stesso sistema biologico (es.Emofilia A e B) Eterogeneità genetica non allelica: mutazioni di pt # coinvolte nello stesso sistema biologico (es.Emofilia A e B) Pleiotropia: effetti fenotipici # in organi # (es. Carenza di insulina sui # metabolismi) Pleiotropia: effetti fenotipici # in organi # (es. Carenza di insulina sui # metabolismi)
; Eterogeneità genetica allelica: mutazioni#, effetti= sulla stessa pt. (es.mutazioni della G6P deH); Eterogeneità genetica non allelica: mutazioni di pt # coinvolte nello stesso sistema biologico (es.Emofilia A e B) Eterogeneità genetica non allelica: mutazioni di pt # coinvolte nello stesso sistema biologico (es.Emofilia A e B) Pleiotropia: effetti fenotipici # in organi # (es. Carenza di insulina sui # metabolismi) Pleiotropia: effetti fenotipici # in organi # (es. Carenza di insulina sui # metabolismi).")
34
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni geniche:Mutazioni puntiformi Missense: sostituzione di un AA (es. FV Leiden, codone della catena della globina nell anemia falciforme). Missense: sostituzione di un AA (es. FV Leiden, codone della catena della globina nell anemia falciforme). Nonsense: codone di stop ( talassemia). Nonsense: codone di stop ( talassemia). Delezione: di una tripletta (fibrosi cistica manca la PHE nella pt che regola il trasporto del Cl - ). Delezione: di una tripletta (fibrosi cistica manca la PHE nella pt che regola il trasporto del Cl - ). Frameshift: diverso schema di lettura (ins. o del. di 1 o 2 b.p.) (es. Tay-Sachs con modifica dell esosaminidasi A). Frameshift: diverso schema di lettura (ins. o del. di 1 o 2 b.p.) (es. Tay-Sachs con modifica dell esosaminidasi A).
. Missense: sostituzione di un AA (es. FV Leiden, codone della catena della globina nell anemia falciforme). Nonsense: codone di stop ( talassemia). Nonsense: codone di stop ( talassemia). Delezione: di una tripletta (fibrosi cistica manca la PHE nella pt che regola il trasporto del Cl - ). Delezione: di una tripletta (fibrosi cistica manca la PHE nella pt che regola il trasporto del Cl - ). Frameshift: diverso schema di lettura (ins. o del. di 1 o 2 b.p.) (es. Tay-Sachs con modifica dell esosaminidasi A). Frameshift: diverso schema di lettura (ins. o del. di 1 o 2 b.p.) (es. Tay-Sachs con modifica dell esosaminidasi A)..")
35
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni silenti Riguardano le regioni non tradotte. Riguardano le regioni non tradotte. Permettono lincorporazione dello stesso AA Permettono lincorporazione dello stesso AA Neutre: producono AA biologicamente equivalenti. Neutre: producono AA biologicamente equivalenti. Possono riguardare i siti di montaggio (Splicing) o di regolazione dell espressione genica (TATA box, poli A) Possono riguardare i siti di montaggio (Splicing) o di regolazione dell espressione genica (TATA box, poli A)
 o di regolazione dell espressione genica (TATA box, poli A) Possono riguardare i siti di montaggio (Splicing) o di regolazione dell espressione genica (TATA box, poli A).")
36
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Mutazioni per amplificazione Malattie da triplette Malattie da triplette Associate a malattie neurodegenerative (atrofia muscolare spino bulbare). Associate a malattie neurodegenerative (atrofia muscolare spino bulbare). Sindrome dell X fragile (ritardo mentale) ; Sindrome dell X fragile (ritardo mentale) ; Distrofia miotonica (distrofia muscolare, miotonia, aritmie). Distrofia miotonica (distrofia muscolare, miotonia, aritmie).
. Sindrome dell X fragile (ritardo mentale) ; Sindrome dell X fragile (ritardo mentale) ; Distrofia miotonica (distrofia muscolare, miotonia, aritmie). Distrofia miotonica (distrofia muscolare, miotonia, aritmie)..")
37
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Sindrome dell X fragile Una parte del comatide è attaccato al cromosoma da un sottile filo di DNA: sito fragile Una parte del comatide è attaccato al cromosoma da un sottile filo di DNA: sito fragile La posizione dei siti fragili è conservata nelle cellule di un individuo e di una famiglia La posizione dei siti fragili è conservata nelle cellule di un individuo e di una famiglia Nell X fragile la tripletta CGG è ripetuta da 200 a 1000 volte (50 nell X normale) Nell X fragile la tripletta CGG è ripetuta da 200 a 1000 volte (50 nell X normale) La sindrome, più evidente nei maschi comporta ritardo mentale La sindrome, più evidente nei maschi comporta ritardo mentale E alterata la funzionalità di un gene che codifica per la Proteina del Ritardo Mentale da X Fragile (FMRP) E alterata la funzionalità di un gene che codifica per la Proteina del Ritardo Mentale da X Fragile (FMRP)
 Nell X fragile la tripletta CGG è ripetuta da 200 a 1000 volte (50 nell X normale) La sindrome, più evidente nei maschi comporta ritardo mentale La sindrome, più evidente nei maschi comporta ritardo mentale E alterata la funzionalità di un gene che codifica per la Proteina del Ritardo Mentale da X Fragile (FMRP) E alterata la funzionalità di un gene che codifica per la Proteina del Ritardo Mentale da X Fragile (FMRP)")
38
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE GENETICHE Malattie genomiche e cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie genomiche e cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile.
. Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile..")
39
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE MENDELIANE O MONOGENICHE Più di 4500 malattie causate da alterazione di un singolo gene che si trasmettono con modalità autosomica dominante, autosomica recessiva o legate al sesso (dominanti o recesssive). Più di 4500 malattie causate da alterazione di un singolo gene che si trasmettono con modalità autosomica dominante, autosomica recessiva o legate al sesso (dominanti o recesssive). 85% di mutazioni familiari, 15% de novo 85% di mutazioni familiari, 15% de novo PROBANDO: membro di una famiglia a causa del quale si analizza la malattia PROBANDO: membro di una famiglia a causa del quale si analizza la malattia
. 85% di mutazioni familiari, 15% de novo 85% di mutazioni familiari, 15% de novo PROBANDO: membro di una famiglia a causa del quale si analizza la malattia PROBANDO: membro di una famiglia a causa del quale si analizza la malattia.")
40
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE DOMINANTI(I) Basta un solo allele mutato per avere la malattia. Aa e aa sono malati, ma aa in genere muore in utero. Basta un solo allele mutato per avere la malattia. Aa e aa sono malati, ma aa in genere muore in utero. e trasmettono la malattia. Se un solo genitore é malato (generalmente Aa), 50% dei figli é sano, 50% dei figli é malato. e trasmettono la malattia. Se un solo genitore é malato (generalmente Aa), 50% dei figli é sano, 50% dei figli é malato.
, 50% dei figli é sano, 50% dei figli é malato. e trasmettono la malattia. Se un solo genitore é malato (generalmente Aa), 50% dei figli é sano, 50% dei figli é malato..")
41
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE DOMINANTI(II) PENETRANZA: frequenza con cui i soggetti portatori del gene mutato esprimono la malattia (in genere < 100%) PENETRANZA: frequenza con cui i soggetti portatori del gene mutato esprimono la malattia (in genere < 100%) ESPRESSIVTA VARIABILE: livello di gravità con cui una malattia autosomica dominante si esprime. ESPRESSIVTA VARIABILE: livello di gravità con cui una malattia autosomica dominante si esprime. Comparsa ± tardiva della sintomatologia. Comparsa ± tardiva della sintomatologia. Nell albero genealogico: verticalità della trasmissione (ogni malato ha un genitore malato) Nell albero genealogico: verticalità della trasmissione (ogni malato ha un genitore malato) Biological fitness: quanto la malattia interferisce con la capacità riproduttiva. Biological fitness: quanto la malattia interferisce con la capacità riproduttiva.
 Nell albero genealogico: verticalità della trasmissione (ogni malato ha un genitore malato) Biological fitness: quanto la malattia interferisce con la capacità riproduttiva. Biological fitness: quanto la malattia interferisce con la capacità riproduttiva..")
42
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE DOMINANTI(III) Von Willebrand (1:125) Von Willebrand (1:125) Ipercolesterolemia famliare (1:500) Ipercolesterolemia famliare (1:500) Policistosi renale dell adulto (1: 1250) Policistosi renale dell adulto (1: 1250) Stenosi subaortica idiopatica ipertrofica (1:1500) Stenosi subaortica idiopatica ipertrofica (1:1500) Neurofibromatosi(1:3000) Neurofibromatosi(1:3000) Poliposi familiare (1:8000) Poliposi familiare (1:8000) Sindrome di Marfan (1:20000)(proporzioni eunucoidi, aracnodattilia, malformazioni oculari e cardivascolari) Sindrome di Marfan (1:20000)(proporzioni eunucoidi, aracnodattilia, malformazioni oculari e cardivascolari)
 Von Willebrand (1:125) Von Willebrand (1:125) Ipercolesterolemia famliare (1:500) Ipercolesterolemia famliare (1:500) Policistosi renale dell adulto (1: 1250) Policistosi renale dell adulto (1: 1250) Stenosi subaortica idiopatica ipertrofica (1:1500) Stenosi subaortica idiopatica ipertrofica (1:1500) Neurofibromatosi(1:3000) Neurofibromatosi(1:3000) Poliposi familiare (1:8000) Poliposi familiare (1:8000) Sindrome di Marfan (1:20000)(proporzioni eunucoidi, aracnodattilia, malformazioni oculari e cardivascolari) Sindrome di Marfan (1:20000)(proporzioni eunucoidi, aracnodattilia, malformazioni oculari e cardivascolari)")
43
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE RECESSIVE(I) Per esprimere la malattia sono necessarie condizioni di omozigosi degli alleli mutati (aa) Per esprimere la malattia sono necessarie condizioni di omozigosi degli alleli mutati (aa) Entrambi i genitori devono essere almeno eterozigoti (Aa) e danno Entrambi i genitori devono essere almeno eterozigoti (Aa) e danno 25% di figli malati (aa) 25% di figli malati (aa) 50% di figli portatori Aa) 50% di figli portatori Aa) 25% di figli sani (AA) 25% di figli sani (AA)
 Per esprimere la malattia sono necessarie condizioni di omozigosi degli alleli mutati (aa) Per esprimere la malattia sono necessarie condizioni di omozigosi degli alleli mutati (aa) Entrambi i genitori devono essere almeno eterozigoti (Aa) e danno Entrambi i genitori devono essere almeno eterozigoti (Aa) e danno 25% di figli malati (aa) 25% di figli malati (aa) 50% di figli portatori Aa) 50% di figli portatori Aa) 25% di figli sani (AA) 25% di figli sani (AA)")
44
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE RECESSIVE(II) La comparsa della malattia in una famiglia dipende: La comparsa della malattia in una famiglia dipende: -dalla frequenza del gene mutato in quella popolazione -Dai matrimoni fra consanguinei Penetranza totale (100%) Mancano di espressività variabile Esordio in età giovanile Nell albero genealogico: orizzontalità della trasmissione (ogni malato ha due genitori sani)
 La comparsa della malattia in una famiglia dipende: La comparsa della malattia in una famiglia dipende: -dalla frequenza del gene mutato in quella popolazione -Dai matrimoni fra consanguinei Penetranza totale (100%) Mancano di espressività variabile Esordio in età giovanile Nell albero genealogico: orizzontalità della trasmissione (ogni malato ha due genitori sani)")
45
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE RECESSIVE(III) Molte riguardano errori genetici del metabolismo dovuti a carenze enzimatiche (accumulo del substrato * e carenza del prodotto della reazione**): Molte riguardano errori genetici del metabolismo dovuti a carenze enzimatiche (accumulo del substrato * e carenza del prodotto della reazione**): * Fenilchetonuria (carenza della fenilalanina idrossilasi). L accumulo di PHE che non viene idrossilata a TIR porta a mielinizzzazione anomala e danni al SNC. **Albinismo (carenza di tirosinasi nei melanosomi): si riduce la sintesi a partire dalla tirosina della Dopa, che é il substrato per la sintesi della melanina.
: si riduce la sintesi a partire dalla tirosina della Dopa, che é il substrato per la sintesi della melanina..")
46
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE RECESSIVE(VI) Emocromatosi (1:200):2 mutazioni missense(Ch6).Difettosa captazione di ferro Emocromatosi (1:200):2 mutazioni missense(Ch6).Difettosa captazione di ferro Fibrosi cistica (1: 2500 caucasici): Il gene sul Ch 7 codifica per una pt. (canale del Cl - ) regolato da fosforilazione cAMP dipendente. Nella Cistic Fibrosis Transmembrane Regulator con Del di 3 basi, manca in 508 PHE e si ha degradazione intracellulare. Fibrosi cistica (1: 2500 caucasici): Il gene sul Ch 7 codifica per una pt. (canale del Cl - ) regolato da fosforilazione cAMP dipendente. Nella Cistic Fibrosis Transmembrane Regulator con Del di 3 basi, manca in 508 PHE e si ha degradazione intracellulare. Ridotte secrezioni di Cl -,Na +, H 2 O,secrezioni dense, danni multisistemici (respiratori, gastrointestinali, pancreas, riproduttivi). Cl - nel sudore >60 mEq/L. Ridotte secrezioni di Cl -,Na +, H 2 O,secrezioni dense, danni multisistemici (respiratori, gastrointestinali, pancreas, riproduttivi). Cl - nel sudore >60 mEq/L.
 regolato da fosforilazione cAMP dipendente. Nella Cistic Fibrosis Transmembrane Regulator con Del di 3 basi, manca in 508 PHE e si ha degradazione intracellulare. Fibrosi cistica (1: 2500 caucasici): Il gene sul Ch 7 codifica per una pt. (canale del Cl - ) regolato da fosforilazione cAMP dipendente. Nella Cistic Fibrosis Transmembrane Regulator con Del di 3 basi, manca in 508 PHE e si ha degradazione intracellulare. Ridotte secrezioni di Cl -,Na +, H 2 O,secrezioni dense, danni multisistemici (respiratori, gastrointestinali, pancreas, riproduttivi). Cl - nel sudore >60 mEq/L. Ridotte secrezioni di Cl -,Na +, H 2 O,secrezioni dense, danni multisistemici (respiratori, gastrointestinali, pancreas, riproduttivi). Cl - nel sudore >60 mEq/L..")
47
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

48
MALATTIE AUTOSOMICHE RECESSIVE(V) Glicogenosi:(1:50000).Malattie ereditarie da carenza di enzimi del metabolismo del glicogeno. Glicogenosi:(1:50000).Malattie ereditarie da carenza di enzimi del metabolismo del glicogeno. Malattia di Von Gierke: assenza di glucoso 6-fosatasi.Forma epatica,ipoglicemia. Malattia di Von Gierke: assenza di glucoso 6-fosatasi.Forma epatica,ipoglicemia. Malattia di Pompe: assenza di maltasi (Glu-Glu) lisosomiale. Miopatia anche cardiaca. Malattia di Pompe: assenza di maltasi (Glu-Glu) lisosomiale. Miopatia anche cardiaca. Malattia di McArdle: assenza di fosforilasi muscolare.Fatica muscolare, rabdomiolisi, mioglobinuria Malattia di McArdle: assenza di fosforilasi muscolare.Fatica muscolare, rabdomiolisi, mioglobinuria
.Malattie ereditarie da carenza di enzimi del metabolismo del glicogeno. Malattia di Von Gierke: assenza di glucoso 6-fosatasi.Forma epatica,ipoglicemia. Malattia di Von Gierke: assenza di glucoso 6-fosatasi.Forma epatica,ipoglicemia. Malattia di Pompe: assenza di maltasi (Glu-Glu) lisosomiale. Miopatia anche cardiaca. Malattia di Pompe: assenza di maltasi (Glu-Glu) lisosomiale. Miopatia anche cardiaca. Malattia di McArdle: assenza di fosforilasi muscolare.Fatica muscolare, rabdomiolisi, mioglobinuria Malattia di McArdle: assenza di fosforilasi muscolare.Fatica muscolare, rabdomiolisi, mioglobinuria.")
49
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE AUTOSOMICHE RECESSIVE(VI) Malattie da accumulo intralisosomiale: mancano enzimi degradativi e si accumulano i substrati nei lisosomi. Malattie da accumulo intralisosomiale: mancano enzimi degradativi e si accumulano i substrati nei lisosomi. Sfingolipidi, sfingomielina (Niemann-Pick), gangliosidi (Tay-Sachs), glucocerebrosidi (Gaucher), galattocerebrosidi (Krabbe), mucopolisaccaridi (Hurler) o glicosaminoglicani. Sfingolipidi, sfingomielina (Niemann-Pick), gangliosidi (Tay-Sachs), glucocerebrosidi (Gaucher), galattocerebrosidi (Krabbe), mucopolisaccaridi (Hurler) o glicosaminoglicani. Alterazioni nervose, epatosplenomegalia, alterazioni scheletriche. Alterazioni nervose, epatosplenomegalia, alterazioni scheletriche.
, gangliosidi (Tay-Sachs), glucocerebrosidi (Gaucher), galattocerebrosidi (Krabbe), mucopolisaccaridi (Hurler) o glicosaminoglicani. Sfingolipidi, sfingomielina (Niemann-Pick), gangliosidi (Tay-Sachs), glucocerebrosidi (Gaucher), galattocerebrosidi (Krabbe), mucopolisaccaridi (Hurler) o glicosaminoglicani. Alterazioni nervose, epatosplenomegalia, alterazioni scheletriche. Alterazioni nervose, epatosplenomegalia, alterazioni scheletriche..")
50
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 SINDROMI TALASSEMICHE (AUTOSOMICHE RECESSIVE) Disordini ereditari dovuti ad alterazioni genetiche eterogenee che determinano labolizione o la riduzione della sintesi di una o più catene globiniche Disordini ereditari dovuti ad alterazioni genetiche eterogenee che determinano labolizione o la riduzione della sintesi di una o più catene globiniche Sulla base delle catene mancanti si distinguono α-talassemie, β-talassemie, Sulla base delle catene mancanti si distinguono α-talassemie, β-talassemie, δ-talassemie
 Disordini ereditari dovuti ad alterazioni genetiche eterogenee che determinano labolizione o la riduzione della sintesi di una o più catene globiniche Disordini ereditari dovuti ad alterazioni genetiche eterogenee che determinano labolizione o la riduzione della sintesi di una o più catene globiniche Sulla base delle catene mancanti si distinguono α-talassemie, β-talassemie, Sulla base delle catene mancanti si distinguono α-talassemie, β-talassemie, δ-talassemie")
51
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 GENI GLOBINICI Sul Ch 11 sono localizzati i geni β simili : β, ε, γ, δ Sul Ch 11 sono localizzati i geni β simili : β, ε, γ, δ Sul Ch 16 sono localizzati i geni α simili : ζ, α Sul Ch 16 sono localizzati i geni α simili : ζ, α Nell adulto esiste HbA (α2β2) ed HbA2 (α2δ2) Nell adulto esiste HbA (α2β2) ed HbA2 (α2δ2) Nel feto esiste HbA2 (α2δ2)e HbF (α2γ2) Nel feto esiste HbA2 (α2δ2)e HbF (α2γ2)
 ed HbA2 (α2δ2) Nell adulto esiste HbA (α2β2) ed HbA2 (α2δ2) Nel feto esiste HbA2 (α2δ2)e HbF (α2γ2) Nel feto esiste HbA2 (α2δ2)e HbF (α2γ2)")
52
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 α- TALASSEMIA I geni per lα-globina sono quattro (α1 e α2) I geni per lα-globina sono quattro (α1 e α2) Nelle α talassemie si ha delezione di geni α globinici: nelle forme (--/--) talassemia la delezione è molto ampia ed interessa sia i geni α1 che i geni α2 (assenza completa di sintesi delle catene α); Nelle α talassemie si ha delezione di geni α globinici: nelle forme (--/--) talassemia la delezione è molto ampia ed interessa sia i geni α1 che i geni α2 (assenza completa di sintesi delle catene α); Nelle forme (-α/α α) (--/α α) (-α/-α) (--/-α)- talassemia viene risparmiato almeno un gene α globinico (ridotta sintesi delle catene α) Nelle forme (-α/α α) (--/α α) (-α/-α) (--/-α)- talassemia viene risparmiato almeno un gene α globinico (ridotta sintesi delle catene α)
 I geni per lα-globina sono quattro (α1 e α2) Nelle α talassemie si ha delezione di geni α globinici: nelle forme (--/--) talassemia la delezione è molto ampia ed interessa sia i geni α1 che i geni α2 (assenza completa di sintesi delle catene α); Nelle α talassemie si ha delezione di geni α globinici: nelle forme (--/--) talassemia la delezione è molto ampia ed interessa sia i geni α1 che i geni α2 (assenza completa di sintesi delle catene α); Nelle forme (-α/α α) (--/α α) (-α/-α) (--/-α)- talassemia viene risparmiato almeno un gene α globinico (ridotta sintesi delle catene α) Nelle forme (-α/α α) (--/α α) (-α/-α) (--/-α)- talassemia viene risparmiato almeno un gene α globinico (ridotta sintesi delle catene α)")
53
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 α- TALASSEMIA: forme cliniche (I) IDROPE FETALE (--/--) : IDROPE FETALE (--/--) : sindrome caratterizzata da morte intrauterina del feto o morte entro poche ore del feto nato a termine. E dovuta ad assenza totale delle catene α.

54
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 α- TALASSEMIA: forme cliniche (II) MALATTIA DA HbH (--/-α) : trasmissione di uno solo dei quattro geni α- globinici con conseguente grave riduzione della sintesi di questa catena. Quasi tutta l Hb è HbH, formata da 4 catene β. Si hanno precipitati di questa Hb in circolo (corpi di Heinz). MALATTIA DA HbH (--/-α) : trasmissione di uno solo dei quattro geni α- globinici con conseguente grave riduzione della sintesi di questa catena. Quasi tutta l Hb è HbH, formata da 4 catene β. Si hanno precipitati di questa Hb in circolo (corpi di Heinz). Sempre presenza di anemia (Hb=7-10 gr/dl) con quadro clinico di talassemia intermedia Sempre presenza di anemia (Hb=7-10 gr/dl) con quadro clinico di talassemia intermedia Ittero, splenomegalia, ulcere malleolari. Ittero, splenomegalia, ulcere malleolari.
. MALATTIA DA HbH (--/-α) : trasmissione di uno solo dei quattro geni α- globinici con conseguente grave riduzione della sintesi di questa catena. Quasi tutta l Hb è HbH, formata da 4 catene β. Si hanno precipitati di questa Hb in circolo (corpi di Heinz). Sempre presenza di anemia (Hb=7-10 gr/dl) con quadro clinico di talassemia intermedia Sempre presenza di anemia (Hb=7-10 gr/dl) con quadro clinico di talassemia intermedia Ittero, splenomegalia, ulcere malleolari. Ittero, splenomegalia, ulcere malleolari..")
55
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 α- TALASSEMIA: forme cliniche (III) α-TALASSEMIA MINOR: (--/α α) (-α/- α): Delezione di 2 dei 4 geni globinici. Clinicamente silente. α-TALASSEMIA MINOR: (--/α α) (-α/- α): Delezione di 2 dei 4 geni globinici. Clinicamente silente. Lemocromo mostra Hb normale o lievemente ridotta, incremento dei globuli rossi microcitemici FORMA SILENTE: (-α/α α)
 (-α/- α): Delezione di 2 dei 4 geni globinici. Clinicamente silente. Lemocromo mostra Hb normale o lievemente ridotta, incremento dei globuli rossi microcitemici FORMA SILENTE: (-α/α α).")
56
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 β-TALASSEMIE Condizioni patologiche caratterizzate dalla diminuita (β+ talassemia) o assente (β0 talassemia) sintesi delle catene β-globiniche. Condizioni patologiche caratterizzate dalla diminuita (β+ talassemia) o assente (β0 talassemia) sintesi delle catene β-globiniche. Le lesioni genetiche sono molto eterogenee: mutazioni puntiformi non senso; frame shift; mutazioni di sequenze introniche con alterazione di splicing; mutazioni del promoter con riduzione di sintesi globinica Le lesioni genetiche sono molto eterogenee: mutazioni puntiformi non senso; frame shift; mutazioni di sequenze introniche con alterazione di splicing; mutazioni del promoter con riduzione di sintesi globinica
 o assente (β0 talassemia) sintesi delle catene β-globiniche. Le lesioni genetiche sono molto eterogenee: mutazioni puntiformi non senso; frame shift; mutazioni di sequenze introniche con alterazione di splicing; mutazioni del promoter con riduzione di sintesi globinica Le lesioni genetiche sono molto eterogenee: mutazioni puntiformi non senso; frame shift; mutazioni di sequenze introniche con alterazione di splicing; mutazioni del promoter con riduzione di sintesi globinica.")
57
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 β- TALASSEMIA: forme cliniche (I) Talassemia maior : (CONDIZIONE DI OMOZIGOSI) sindrome caratterizzata da variabilità nellespressione clinica legata alleterogeneità delle lesioni genetiche (forme β0/β0, forme β0β+, forme β+β+ con % diversa di HbF (αγ), HbA (αß), HbA2 (αδ) Talassemia maior : (CONDIZIONE DI OMOZIGOSI) sindrome caratterizzata da variabilità nellespressione clinica legata alleterogeneità delle lesioni genetiche (forme β0/β0, forme β0β+, forme β+β+ con % diversa di HbF (αγ), HbA (αß), HbA2 (αδ) Patogenesi : anemia grave ad insorgenza postnatale da eritropoiesi inefficace ed emolisi periferica; epatosplenomegalia, ittero, cardiomegalia, alterazioni ossee (facies microcitemica), ritardo dello sviluppo corporeo, disturbi endocrini, ulcere cutanee (insufficienza venosa cronica) Patogenesi : anemia grave ad insorgenza postnatale da eritropoiesi inefficace ed emolisi periferica; epatosplenomegalia, ittero, cardiomegalia, alterazioni ossee (facies microcitemica), ritardo dello sviluppo corporeo, disturbi endocrini, ulcere cutanee (insufficienza venosa cronica)
 Talassemia maior : (CONDIZIONE DI OMOZIGOSI) sindrome caratterizzata da variabilità nellespressione clinica legata alleterogeneità delle lesioni genetiche (forme β0/β0, forme β0β+, forme β+β+ con % diversa di HbF (αγ), HbA (αß), HbA2 (αδ) Talassemia maior : (CONDIZIONE DI OMOZIGOSI) sindrome caratterizzata da variabilità nellespressione clinica legata alleterogeneità delle lesioni genetiche (forme β0/β0, forme β0β+, forme β+β+ con % diversa di HbF (αγ), HbA (αß), HbA2 (αδ) Patogenesi : anemia grave ad insorgenza postnatale da eritropoiesi inefficace ed emolisi periferica; epatosplenomegalia, ittero, cardiomegalia, alterazioni ossee (facies microcitemica), ritardo dello sviluppo corporeo, disturbi endocrini, ulcere cutanee (insufficienza venosa cronica) Patogenesi : anemia grave ad insorgenza postnatale da eritropoiesi inefficace ed emolisi periferica; epatosplenomegalia, ittero, cardiomegalia, alterazioni ossee (facies microcitemica), ritardo dello sviluppo corporeo, disturbi endocrini, ulcere cutanee (insufficienza venosa cronica)")
58
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 β- TALASSEMIA: forme cliniche (II) Talassemia maior :decorso, prognosi, terapia Talassemia maior :decorso, prognosi, terapia La malattia lasciata a sé è mortale entro pochi mesi di vita. La terapia trasfusionale fa sopravvivere fino alla 4°-5° decade. La malattia lasciata a sé è mortale entro pochi mesi di vita. La terapia trasfusionale fa sopravvivere fino alla 4°-5° decade. Le complicanze legate alla pratica trasfusionale riguardano l emocromatosi. Le complicanze legate alla pratica trasfusionale riguardano l emocromatosi. Terapia: Regime ipertrasfusionale; terapia ferrochelante (Deferoxamina); splenectomia; TMO Terapia: Regime ipertrasfusionale; terapia ferrochelante (Deferoxamina); splenectomia; TMO
; splenectomia; TMO Terapia: Regime ipertrasfusionale; terapia ferrochelante (Deferoxamina); splenectomia; TMO.")
59
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 β- TALASSEMIA: forme cliniche (III) Talassemia intermedia: ampio spettro di quadri clinici a gravità inferiore rispetto alla maior, geneticamente eterogenei (forme eterozigoti composte) Talassemia intermedia: ampio spettro di quadri clinici a gravità inferiore rispetto alla maior, geneticamente eterogenei (forme eterozigoti composte) Spesso cè una riduzione dello sbilanciamento della sintesi di catene α e ß (mutazioni che provengono una da un genitore e una dall altro). Spesso cè una riduzione dello sbilanciamento della sintesi di catene α e ß (mutazioni che provengono una da un genitore e una dall altro). Clinica: anemia ad esordio più tardivo rispetto alla maior, splenomegalia progressiva, anisopoichilocitosi,iperbilirubinemia Clinica: anemia ad esordio più tardivo rispetto alla maior, splenomegalia progressiva, anisopoichilocitosi,iperbilirubinemia Terapia: Splenectomia, trasfusioni, terapia ferrochelante Terapia: Splenectomia, trasfusioni, terapia ferrochelante
. Clinica: anemia ad esordio più tardivo rispetto alla maior, splenomegalia progressiva, anisopoichilocitosi,iperbilirubinemia Clinica: anemia ad esordio più tardivo rispetto alla maior, splenomegalia progressiva, anisopoichilocitosi,iperbilirubinemia Terapia: Splenectomia, trasfusioni, terapia ferrochelante Terapia: Splenectomia, trasfusioni, terapia ferrochelante.")
60
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 β- TALASSEMIA: forme cliniche (IV) Talassemia minor: condizione asintomatica determinata dallo stato di eterozigosi per il gene della ß0 o ß+ talassemia. Talassemia minor: condizione asintomatica determinata dallo stato di eterozigosi per il gene della ß0 o ß+ talassemia. C è microcitosi compensata da un aumento dei globuli rossi in assenza di anemia. C è microcitosi compensata da un aumento dei globuli rossi in assenza di anemia.

61
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Anemia drepanocitica (I) Anemia emolitica cronica ad eredità autosomica recessiva, caratterizzata dalla morfologia a falce delle emazie dovuta alla presenza di Hb patologica: HbS Anemia emolitica cronica ad eredità autosomica recessiva, caratterizzata dalla morfologia a falce delle emazie dovuta alla presenza di Hb patologica: HbS
 Anemia emolitica cronica ad eredità autosomica recessiva, caratterizzata dalla morfologia a falce delle emazie dovuta alla presenza di Hb patologica: HbS Anemia emolitica cronica ad eredità autosomica recessiva, caratterizzata dalla morfologia a falce delle emazie dovuta alla presenza di Hb patologica: HbS")
62
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Anemia drepanocitica (II) Carattere autosomico recessivo Carattere autosomico recessivo Il 6° AA della catena ß della globina è Val anzicché GLU; l Hb è meno solubile e cristallizza dando agli eritrociti la forma a falce Il 6° AA della catena ß della globina è Val anzicché GLU; l Hb è meno solubile e cristallizza dando agli eritrociti la forma a falce Molto diffusa in Africa (1/12 è eterozigote), rende gli eterozigoti resistenti al Plasmodium malariae Molto diffusa in Africa (1/12 è eterozigote), rende gli eterozigoti resistenti al Plasmodium malariae
 Carattere autosomico recessivo Carattere autosomico recessivo Il 6° AA della catena ß della globina è Val anzicché GLU; l Hb è meno solubile e cristallizza dando agli eritrociti la forma a falce Il 6° AA della catena ß della globina è Val anzicché GLU; l Hb è meno solubile e cristallizza dando agli eritrociti la forma a falce Molto diffusa in Africa (1/12 è eterozigote), rende gli eterozigoti resistenti al Plasmodium malariae Molto diffusa in Africa (1/12 è eterozigote), rende gli eterozigoti resistenti al Plasmodium malariae")
63
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE RECESSIVE LEGATE AL SESSO (I) Sono dovute a geni che mappano sul Ch X. Sono dovute a geni che mappano sul Ch X. Emizigosi obbligata del Eterocromnatizzazione casuale di X nella Emizigosi obbligata del Eterocromnatizzazione casuale di X nella (X*Y)esprime la malattia e trasmette il gene alle figlie (portatrici sane X*X) e non ai figli (XY). (X*Y)esprime la malattia e trasmette il gene alle figlie (portatrici sane X*X) e non ai figli (XY). (X*X) portatrici, trasmettono la malattia al 50% dei figli, mentre il 50% delle figlie é portatrice (X*X) portatrici, trasmettono la malattia al 50% dei figli, mentre il 50% delle figlie é portatrice
esprime la malattia e trasmette il gene alle figlie (portatrici sane X*X) e non ai figli (XY). (X*Y)esprime la malattia e trasmette il gene alle figlie (portatrici sane X*X) e non ai figli (XY). (X*X) portatrici, trasmettono la malattia al 50% dei figli, mentre il 50% delle figlie é portatrice (X*X) portatrici, trasmettono la malattia al 50% dei figli, mentre il 50% delle figlie é portatrice.")
64
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Trasmissione emofilia: Madre portatricePadre sano Femmina portatrice Femmina sana Maschio emofilico Maschio sano XEXE XXY XEXE X XEXE Y XX XY

65
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Trasmissione emofilia: Madre sanaPadre emofilico Femmina portatrice Maschio sano Maschio sano XXXEXE Y XXEXE XY XXEXE XY

66
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 Trasmissione emofilia : Madre portatricePadre emofilico Femmina emofilica Femmina portatrice Maschio emofilico Maschio sano XEXE XXEXE Y XEXE XEXE XEXE YXXEXE XY

67
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE RECESSIVE LEGATE AL SESSO (II) Emofilia A e B Emofilia A e B Daltonismo Daltonismo Carenza di Glu 6-P deH Carenza di Glu 6-P deH Lesch-Nyhan:carenza di ipoxantina- guanina fosforibosil transferasi (iperuricemia, problemi neurologici) Lesch-Nyhan:carenza di ipoxantina- guanina fosforibosil transferasi (iperuricemia, problemi neurologici) X fragile (1:1000): repeat CGG. 2/3 delle hanno ritardo mentale lieve. X fragile (1:1000): repeat CGG. 2/3 delle hanno ritardo mentale lieve.
: repeat CGG. 2/3 delle hanno ritardo mentale lieve..")
68
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE DOMINANTI LEGATE AL SESSO (I) Rarissime (es.rachitismo vitaminico D-resistente ipofosfatemico) Rarissime (es.rachitismo vitaminico D-resistente ipofosfatemico) e esprimono la malattia (meno grave nella eterozigote) e esprimono la malattia (meno grave nella eterozigote) eterozigoti danno 50%figlie malate e 50% figli malati eterozigoti danno 50%figlie malate e 50% figli malati malati trasmettono la malattia a tutte le figlie e non ai figli. malati trasmettono la malattia a tutte le figlie e non ai figli.

69
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE GENETICHE Malattie cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile.
. Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile. Malattie legate ad eredità mitocondriale:si trasmettono solo in linea femminile..")
70
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE CON EREDITARIETA MULTIFATTORIALE (I) Sono causate da mutazioni multiple e da fattori ambientali. Sono causate da mutazioni multiple e da fattori ambientali. Fattori di rischio: sono associati all insorgenza della malattia, ma non ne sono la causa (ognuno ha un suo peso). Fattori di rischio: sono associati all insorgenza della malattia, ma non ne sono la causa (ognuno ha un suo peso). Rischio Relativo: di un soggetto tiene conto del rischio genetico e del rischio ambientale Rischio Relativo: di un soggetto tiene conto del rischio genetico e del rischio ambientale
. Fattori di rischio: sono associati all insorgenza della malattia, ma non ne sono la causa (ognuno ha un suo peso). Rischio Relativo: di un soggetto tiene conto del rischio genetico e del rischio ambientale Rischio Relativo: di un soggetto tiene conto del rischio genetico e del rischio ambientale.")
71
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE CON EREDITARIETA MULTIFATTORIALE (II) Il rischio di un malato di trasmettere la malattia alla prole dipende dal numero di geni mutati (ignoto) in loci indipendenti i cui effetti interagiscono in modo cumulativo (additivo o moltiplicativo) Il rischio di un malato di trasmettere la malattia alla prole dipende dal numero di geni mutati (ignoto) in loci indipendenti i cui effetti interagiscono in modo cumulativo (additivo o moltiplicativo) La probabilità dipende dal numero di parenti malati, dalla gravità della malattia, dalla genetica della popolazione e dalla diversa prevalenza La probabilità dipende dal numero di parenti malati, dalla gravità della malattia, dalla genetica della popolazione e dalla diversa prevalenza
 Il rischio di un malato di trasmettere la malattia alla prole dipende dal numero di geni mutati (ignoto) in loci indipendenti i cui effetti interagiscono in modo cumulativo (additivo o moltiplicativo) Il rischio di un malato di trasmettere la malattia alla prole dipende dal numero di geni mutati (ignoto) in loci indipendenti i cui effetti interagiscono in modo cumulativo (additivo o moltiplicativo) La probabilità dipende dal numero di parenti malati, dalla gravità della malattia, dalla genetica della popolazione e dalla diversa prevalenza La probabilità dipende dal numero di parenti malati, dalla gravità della malattia, dalla genetica della popolazione e dalla diversa prevalenza")
72
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE CON EREDITARIETA MULTIFATTORIALE (III) Labbro leporino Labbro leporino Diabete mellito di tipo II Diabete mellito di tipo II Malattie depressive Malattie depressive Ipertensione Ipertensione Ritardo mentale Ritardo mentale Difetti di chiusura del tubo neurale Difetti di chiusura del tubo neurale
 Labbro leporino Labbro leporino Diabete mellito di tipo II Diabete mellito di tipo II Malattie depressive Malattie depressive Ipertensione Ipertensione Ritardo mentale Ritardo mentale Difetti di chiusura del tubo neurale Difetti di chiusura del tubo neurale")
73
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE GENETICHE Malattie cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie cromosomiche: Aneuploidie (±Ch). Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale: Malattie legate ad eredità mitocondriale: si trasmettono solo in linea femminile.
. Lesioni visibili al microscopio. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie monogeniche: Trasmesse con meccanismo Mendeliano. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie multifattoriali: Causate da interazioni fra diversi fattori genetici ed ambientali. Malattie legate ad eredità mitocondriale: Malattie legate ad eredità mitocondriale: si trasmettono solo in linea femminile..")
74
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE DEL DNA MITOCONDRIALE(I) Il DNA genomico mitocondriale é circolare, costituito da ~ 16000 b.p. e codifica per gli enzimi della fosforilazione ossidativa. Il DNA genomico mitocondriale é circolare, costituito da ~ 16000 b.p. e codifica per gli enzimi della fosforilazione ossidativa. 2-10 genomi per mitocondrio 2-10 genomi per mitocondrio Uova: 200.000-300.000 copie di mt DNA Uova: 200.000-300.000 copie di mt DNA Spermi: perdono I mitocondri nella fertilizzazione Spermi: perdono I mitocondri nella fertilizzazione Eteroplasmia: coesistono molecole di mtDNA mutate e normali Eteroplasmia: coesistono molecole di mtDNA mutate e normali

75
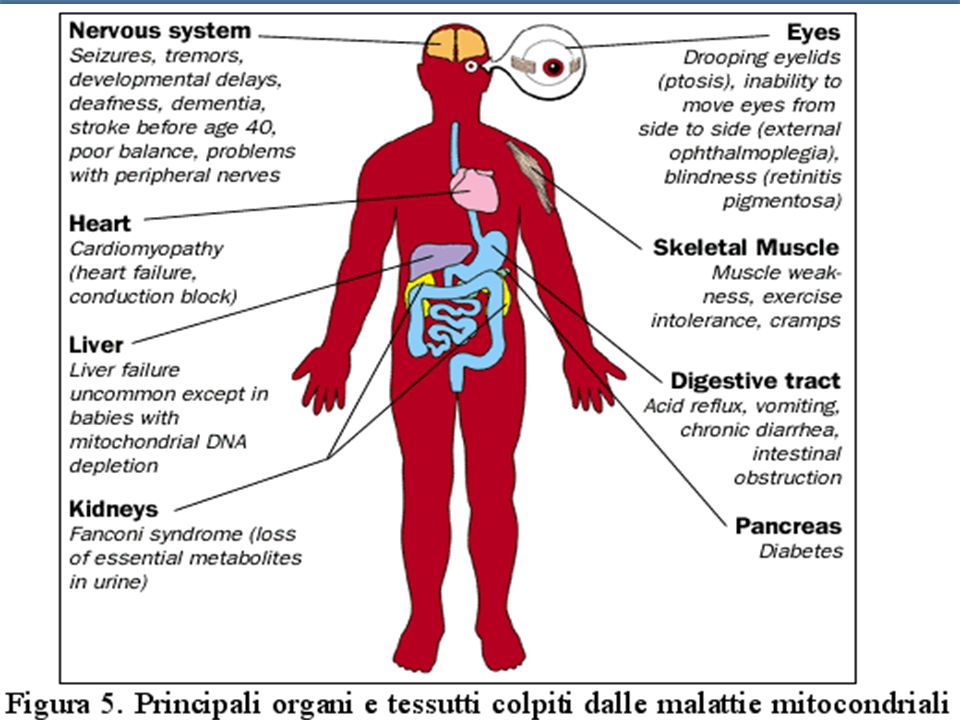
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 MALATTIE DEL DNA MITOCONDRIALE(II ) Mutazioni puntiformi o delezioni. Mutazioni puntiformi o delezioni. malata può trasmettere la malattia a tutti I figli ( e ). La mutazione é nella cellula uovo. malata può trasmettere la malattia a tutti I figli ( e ). La mutazione é nella cellula uovo. malato non trasmette la malattia alla progenie. Lo spermatozoo perde i mitocondri malato non trasmette la malattia alla progenie. Lo spermatozoo perde i mitocondri Malattie progressive dei muscoli, cuore, SNC (es. Neuropatia ottica ereditaria di Leber) Malattie progressive dei muscoli, cuore, SNC (es. Neuropatia ottica ereditaria di Leber)
. La mutazione é nella cellula uovo. malata può trasmettere la malattia a tutti I figli ( e ). La mutazione é nella cellula uovo. malato non trasmette la malattia alla progenie. Lo spermatozoo perde i mitocondri malato non trasmette la malattia alla progenie. Lo spermatozoo perde i mitocondri Malattie progressive dei muscoli, cuore, SNC (es. Neuropatia ottica ereditaria di Leber) Malattie progressive dei muscoli, cuore, SNC (es. Neuropatia ottica ereditaria di Leber).")
76
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

79
Principali malattie da mitDNA MELAS:Miopatia mitocondriale- encefalopatia-acidosi lattica MELAS:Miopatia mitocondriale- encefalopatia-acidosi lattica MND: Motor Neurone desease MND: Motor Neurone desease MEERF:Myoclonic Epilepsy with Ragged Red Fibers MEERF:Myoclonic Epilepsy with Ragged Red Fibers

80
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 DIAGNOSI PRENATALE DELLE MALATTIE GENETICHE (I) Anamnesi: provenienza della famiglia, parentela fra I genitori, malattie nella famiglia. Anamnesi: provenienza della famiglia, parentela fra I genitori, malattie nella famiglia. Cariotipo: le cellule si stimolano in mitosi con fitoemoagglutinina (PHA), si arrestano in metafase con colchicina, si bandeggiano con Giemsa o Chinacrina. Cariotipo: le cellule si stimolano in mitosi con fitoemoagglutinina (PHA), si arrestano in metafase con colchicina, si bandeggiano con Giemsa o Chinacrina.
, si arrestano in metafase con colchicina, si bandeggiano con Giemsa o Chinacrina. Cariotipo: le cellule si stimolano in mitosi con fitoemoagglutinina (PHA), si arrestano in metafase con colchicina, si bandeggiano con Giemsa o Chinacrina..")
81
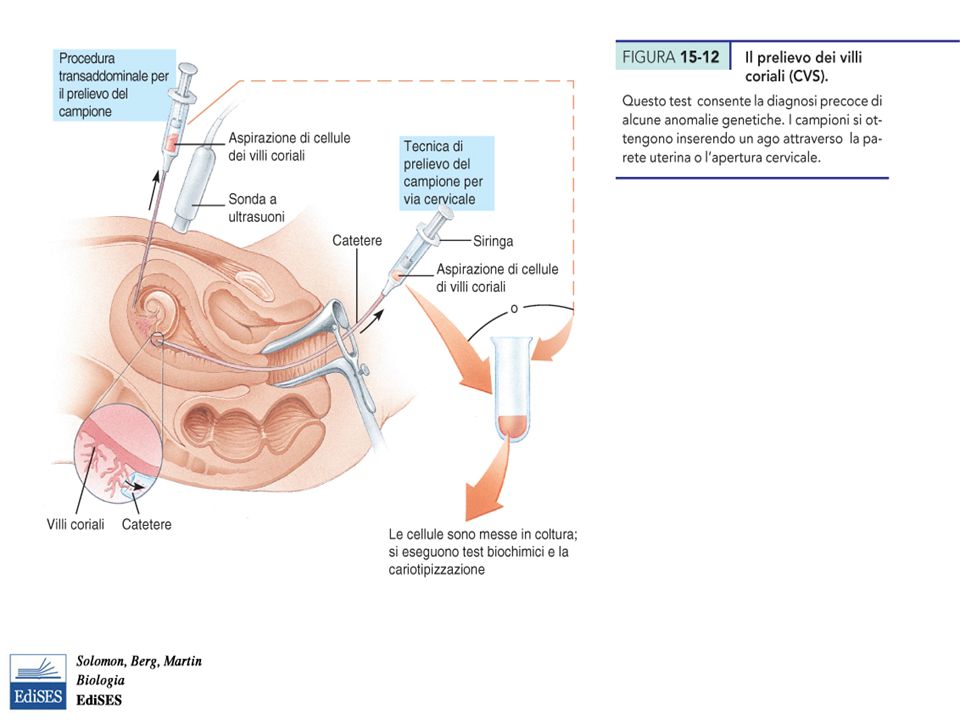
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15 DIAGNOSI PRENATALE DELLE MALATTIE GENETICHE (II) AMNIOCENTESI: 16 settimane. Cariotipo dalle cellule del liquido amniotico. AMNIOCENTESI: 16 settimane. Cariotipo dalle cellule del liquido amniotico. PRELIEVO DEI VILLI CORIALI: 9-12 settimane. Piu rischiosa. PRELIEVO DEI VILLI CORIALI: 9-12 settimane. Piu rischiosa. PRELIEVO DI SANGUE FETALE: 18 settimane. (per emoglobinopatie o isoimmunizzazione) PRELIEVO DI SANGUE FETALE: 18 settimane. (per emoglobinopatie o isoimmunizzazione) ECOGRAFIA ECOGRAFIA TRITEST: AFP, Hcg, Estriolo non coniugato + età materna. Test probabilistco(!) TRITEST: AFP, Hcg, Estriolo non coniugato + età materna. Test probabilistco(!) SPESSORE DELLA PLICA NUCALE: Test probabilistco(!) SPESSORE DELLA PLICA NUCALE: Test probabilistco(!)
 PRELIEVO DI SANGUE FETALE: 18 settimane. (per emoglobinopatie o isoimmunizzazione) ECOGRAFIA ECOGRAFIA TRITEST: AFP, Hcg, Estriolo non coniugato + età materna. Test probabilistco(!) TRITEST: AFP, Hcg, Estriolo non coniugato + età materna. Test probabilistco(!) SPESSORE DELLA PLICA NUCALE: Test probabilistco(!) SPESSORE DELLA PLICA NUCALE: Test probabilistco(!).")
82
Martini – Fondamenti di Anatomia e Fisiologia – Capitolo 15

Presentazioni simili


 Dominanza incompleta>")


 è dominante sul pelo liscio ( r )>")














