Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

 Advanced phase.")
1
LEUCEMIA MIELOIDE CRONICA (CML)
Chronic phase Advanced phase

2
In chronic phase (CP), the bulk of leukaemic stem cells remain capable of undergoing differentiation, leading to the excessive production of mature granulocytes. In advanced phase disease, differentiation has become arrested, probably at the stage of the leukaemia progenitor cell, and the 'aggressive' disease phenotype is caused by the proliferation (self-renewal) of immature blasts. Deleterious genetic events (inset) are believed to accumulate within stem and progenitor cells of the leukaemic clone until there are sufficient secondary mutations to drive the transition from chronic to advanced phase disease. These include: an increase in genomic instability through interference with genomic surveillance and DNA-repair proteins and a progressive telomere shortening. In CP cells essential tumour-suppressor (TS) proteins remain functional and allow cells to undergo replicative senescence or apoptosis. However, in advanced phase blasts there is evidence that TS function has been lost.
 of immature blasts. Deleterious genetic events (inset) are believed to accumulate within stem and progenitor cells of the leukaemic clone until there are sufficient secondary mutations to drive the transition from chronic to advanced phase disease. These include: an increase in genomic instability through interference with genomic surveillance and DNA-repair proteins and a progressive telomere shortening. In CP cells essential tumour-suppressor (TS) proteins remain functional and allow cells to undergo replicative senescence or apoptosis. However, in advanced phase blasts there is evidence that TS function has been lost..")
5
The ABL and BCR proteins
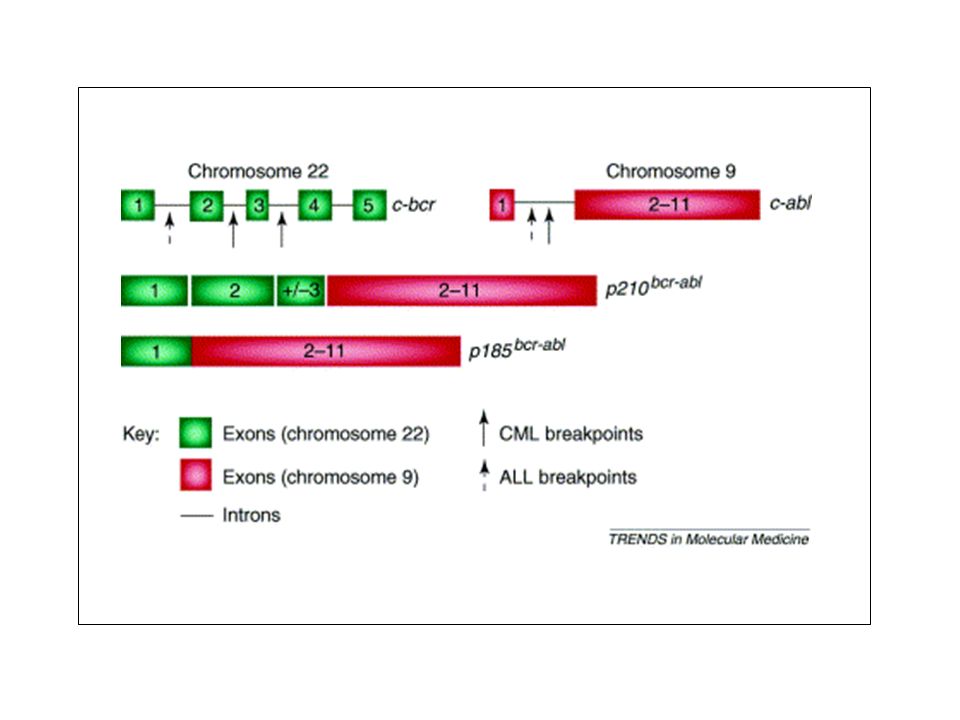
The ABL and BCR proteins. There are several important domains that make up ABL and BCR proteins. a | Two isoforms of ABL (human types 1a and 1b) are generated by alternative splicing of the first exon, one of them (1b) contains a myristoylation modification site (Myr). (Myristoylation is a process that attaches the fourteen-carbon saturated fatty acid myristate to the amino-terminal glycine of proteins.) Apart from the alternatively spliced sequences, the amino-terminal half of ABL contains tandem SRC homology 3 (SH3), SH2 and the tyrosine-kinase (Y-kinase) domains. These domains can assemble into an auto-inhibitory structure, in which the SH3 and SH2 domains function as a 'clamp' that holds the kinase in the 'off' state119, 120. In ABL1b, the myristoyl group at the extreme end of the amino-terminal segment also binds to the tyrosine-kinase domain and functions as a 'latch' that keeps the SH3–SH2 clamp in place119, 121. In its carboxy-terminal region, ABL contains four proline-rich SH3 binding sites (PPs), three nuclear localization signals (NLSs), one nuclear exporting signal (NES), a DNA-binding domain (DBD), and an actin-binding domain (ABD). This actin-binding domain contains binding sites for both monomeric (G) and filamentous (F) forms of actin. The points in ABL that fuses with BCR and GAG (for v-Abl) are indicated. b | BCR contains a coiled-coil (CC) oligomerization domain, a serine/threonine (S/T) kinase domain, a Dbl/CDC24 guanine-nucleotide exchange factor homology (DH) domain and a pleckstrin homology (PH) domain, a putative calcium-dependent lipid binding site (CaLB) and a RAC guanosine triphosphatase-activating protein (RAC-GAP) domain. BCR also contains binding sites for growth factor receptor-bound protein 2 (GRB2) at tyrosine 177 (Y177), as well as for the GRB10, and the ABL proteins, through its SH2 domain. p185, p210 and p230 indicate the points at which BCR most commonly fuses to ABL — these forms are associated with acute lymphoblastic leukaemia, chronic myelogenous leukaemia (CML) and a milder form of CML, respectively. The number of amino acids in each form are indicated in parentheses.
 are generated by alternative splicing of the first exon, one of them (1b) contains a myristoylation modification site (Myr). (Myristoylation is a process that attaches the fourteen-carbon saturated fatty acid myristate to the amino-terminal glycine of proteins.) Apart from the alternatively spliced sequences, the amino-terminal half of ABL contains tandem SRC homology 3 (SH3), SH2 and the tyrosine-kinase (Y-kinase) domains. These domains can assemble into an auto-inhibitory structure, in which the SH3 and SH2 domains function as a clamp that holds the kinase in the off state119, 120. In ABL1b, the myristoyl group at the extreme end of the amino-terminal segment also binds to the tyrosine-kinase domain and functions as a latch that keeps the SH3–SH2 clamp in place119, 121. In its carboxy-terminal region, ABL contains four proline-rich SH3 binding sites (PPs), three nuclear localization signals (NLSs), one nuclear exporting signal (NES), a DNA-binding domain (DBD), and an actin-binding domain (ABD). This actin-binding domain contains binding sites for both monomeric (G) and filamentous (F) forms of actin. The points in ABL that fuses with BCR and GAG (for v-Abl) are indicated. b | BCR contains a coiled-coil (CC) oligomerization domain, a serine/threonine (S/T) kinase domain, a Dbl/CDC24 guanine-nucleotide exchange factor homology (DH) domain and a pleckstrin homology (PH) domain, a putative calcium-dependent lipid binding site (CaLB) and a RAC guanosine triphosphatase-activating protein (RAC-GAP) domain. BCR also contains binding sites for growth factor receptor-bound protein 2 (GRB2) at tyrosine 177 (Y177), as well as for the GRB10, and the ABL proteins, through its SH2 domain. p185, p210 and p230 indicate the points at which BCR most commonly fuses to ABL — these forms are associated with acute lymphoblastic leukaemia, chronic myelogenous leukaemia (CML) and a milder form of CML, respectively. The number of amino acids in each form are indicated in parentheses.")
6
FUNZIONI DI c-Abl Riconoscimento di lesioni a carico del DNA
Progressione del ciclo cellulare Effetti trascrizionali

8
Leukaemogenic signalling of BCR–ABL
Leukaemogenic signalling of BCR–ABL. The p210 form of BCR–ABL is shown in this diagram (see Fig. 2 for fusion point, which occurs after the pleckstrin homology (PH) domain of BCR). The BCR–ABL proteins can form dimers or tetramers through their CC domains, and trans-autophosphorylate (indicated by up and down arrows between protein structures). Phosphorylation at the Y177 residue generates a high-affinity binding site for growth factor receptor-bound protein 2 (GRB2). GRB2 binds to BCR–ABL through its SH2 domain and binds to SOS and GRB2-associated binding protein 2 (GAB2) through its SH3 domains. SOS in turn activates RAS. Following phosphorylation (P) by BCR–ABL, GAB2 recruits phosphatidylinositol 3-kinase (PI3K) and SHP2 proteins. The SH2 domain of ABL can bind SHC, which, following phosphorylation can also recruit GRB2. The ABL SH3 domain and the SH3 binding sites in the carboxy-terminal region can bind several proteins that involve regulations of cell adhesion/migration. Interferon consensus sequence binding protein (ICSBP), also known as interferon regulatory factor 8, negatively regulates proliferation and survival of myeloid cells by inducing differentiation of monocytic cells. JUNB inhibits cell proliferation and survival, partly by antagonizing the RAS downstream target JUN. SIPA1 (signal-induced proliferation-associated gene-1) is a RAP1 GAP that keeps RAP1 inactive. BCR–ABL can promote cell proliferation and survival partly by activating the RAS, SHP2 and PI3K–AKT signalling pathways. It can also downregulate transcription of ICSBP and JUNB, and might also inhibit SIPA1. Red arrows indicate direct interactions and/or activations. Black arrows indicate negative regulations. Broken arrows indicate multiple steps. ABD, actin-binding domain; CC, coiled-coil; DBD, DNA-binding domain; DH, Dbl/CDC24 guanine-nuleotide exchange factor homology; NES, nuclear exporting signal; NLS, nuclear localization signal; PP, proline-rich SH3 binding site; S/T-K, serine/threonine kinase; Y-K, tyrosine kinase.
 domain of BCR). The BCR–ABL proteins can form dimers or tetramers through their CC domains, and trans-autophosphorylate (indicated by up and down arrows between protein structures). Phosphorylation at the Y177 residue generates a high-affinity binding site for growth factor receptor-bound protein 2 (GRB2). GRB2 binds to BCR–ABL through its SH2 domain and binds to SOS and GRB2-associated binding protein 2 (GAB2) through its SH3 domains. SOS in turn activates RAS. Following phosphorylation (P) by BCR–ABL, GAB2 recruits phosphatidylinositol 3-kinase (PI3K) and SHP2 proteins. The SH2 domain of ABL can bind SHC, which, following phosphorylation can also recruit GRB2. The ABL SH3 domain and the SH3 binding sites in the carboxy-terminal region can bind several proteins that involve regulations of cell adhesion/migration. Interferon consensus sequence binding protein (ICSBP), also known as interferon regulatory factor 8, negatively regulates proliferation and survival of myeloid cells by inducing differentiation of monocytic cells. JUNB inhibits cell proliferation and survival, partly by antagonizing the RAS downstream target JUN. SIPA1 (signal-induced proliferation-associated gene-1) is a RAP1 GAP that keeps RAP1 inactive. BCR–ABL can promote cell proliferation and survival partly by activating the RAS, SHP2 and PI3K–AKT signalling pathways. It can also downregulate transcription of ICSBP and JUNB, and might also inhibit SIPA1. Red arrows indicate direct interactions and/or activations. Black arrows indicate negative regulations. Broken arrows indicate multiple steps. ABD, actin-binding domain; CC, coiled-coil; DBD, DNA-binding domain; DH, Dbl/CDC24 guanine-nuleotide exchange factor homology; NES, nuclear exporting signal; NLS, nuclear localization signal; PP, proline-rich SH3 binding site; S/T-K, serine/threonine kinase; Y-K, tyrosine kinase.")
9
BCR-ABL increases the stability of heterogeneous nuclear ribonucleoprotein E2 (HNRNPE2), which binds to a seven nucleotide spacer within the sequence of CEBP mRNA. The binding of HNRNPE2 to CEBP mRNA inhibits the subsequent translation of this transcription factor, leading to the transcriptional downregulation of one of its target genes, CSF3R, which encodes the granulocyte colony-stimulating factor receptor (GCSFR) required for granulocytic differentiation.
 required for granulocytic differentiation..")
10
a | BCR-ABL inhibits the key genome surveillance kinase ataxia telangiectasia and rad 3-related (ATR). BCR-ABL shuttles to the nucleus, in response to a genotoxic insult, where it binds to and inhibits the DNA-damage sensor kinase — ATR. ATR bound by BCR-ABL fails to catalyse the phosphorylation and activation of its substrate, checkpoint kinase 1 (CHK1), which, in turn, fails to disrupt and inactivate the cell division cycle 7 (CDC7)–DBF4 complex. The active CDC7–DBF4 complex promotes the loading of CDC45 into the pre-replication complex (Pre-RC), allowing inappropriate replication to occur. The interaction between CHK1 and the CDC7–DBF4 complex is indicated with a dotted line, as this is a putative function of CHK1 based on the known role of checkpoint kinases in regulating CDC7 and DBF4 in yeast.
, which, in turn, fails to disrupt and inactivate the cell division cycle 7 (CDC7)–DBF4 complex. The active CDC7–DBF4 complex promotes the loading of CDC45 into the pre-replication complex (Pre-RC), allowing inappropriate replication to occur. The interaction between CHK1 and the CDC7–DBF4 complex is indicated with a dotted line, as this is a putative function of CHK1 based on the known role of checkpoint kinases in regulating CDC7 and DBF4 in yeast..")
11
| BCR-ABL interferes with the repair of DNA double-strand breaks (DSBs) by inhibiting the expression of the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs). DNA-PKcs is downregulated in inverse relation to the expression of BCR-ABL in cells. DNA-PKcs binds to the free ends of DSBs, where it forms a complex with the KU70–KU80 heterodimer. Ligation of the broken strands also requires DNA ligase IV, X-ray cross complementation group 4 protein (XRCC4) and the nuclease artemis (not shown).
 and the nuclease artemis (not shown)..")
12
c | BCR-ABL interferes with the repair of DNA double-strand breaks (DSBs) through several effects on the DNA repair protein RAD51. BCR-ABL stimulates the transcription of RAD51 by a mechanism that involves signal transducer and activator of transcription 5 (STAT5). In addition, BCR-ABL activates RAD51 through the phosphorylation of T315 and suppresses its caspase 3-mediated proteolytic degradation (not shown). Homologous recombination (HR) repair of DNA by BCR-ABL-stimulated RAD51 lacks the fidelity of HR repair in untransformed cells. Repair by HR also requires many other proteins, including RAD51 paralogues and the RAD50–MRE11–NBS1 complex (not shown)
. In addition, BCR-ABL activates RAD51 through the phosphorylation of T315 and suppresses its caspase 3-mediated proteolytic degradation (not shown). Homologous recombination (HR) repair of DNA by BCR-ABL-stimulated RAD51 lacks the fidelity of HR repair in untransformed cells. Repair by HR also requires many other proteins, including RAD51 paralogues and the RAD50–MRE11–NBS1 complex (not shown).")
13
PRINCIPALI TERAPIE PER LA CML
idrossiurea interferone- (IFN- ) IFN- + araC trapianto eterologo di cellule staminali trapianto autologo di cellule staminali
 IFN- + araC. trapianto eterologo di cellule staminali. trapianto autologo di cellule staminali.")
14
This highly simplified scheme illustrates some of the more important BCR–ABL signalling pathways. Phosphorylation at the Y177 residue of BCR generates a high-affinity docking site for growth-factor receptor-bound protein 2 (GRB2), which in turn binds the scaffolding adaptor GRB2-associated binding protein 2 (GAB2), as well as SOS (a guanine-nucleotide exchanger of RAS), resulting in RAS–MAPK activation (leading to BCL2 gene transcription). On phosphorylation by BCR–ABL, GAB2 recruits SHP2 (also known as PTPN11) and phosphatidylinositol-3 kinase (PI3K), which leads to AKT activation. BCR–ABL also activates other signalling pathways such as signal transducer and activator of transcription 5 (STAT5), leading to BCLX gene (also known as BCL2L1) transcription and the SRC family kinases (SFKs) LYN and HCK. By contrast, BCR–ABL represses interferon consensus sequence binding protein (ICSBP) transcription through an unknown mechanism, which releases the ISCBP-mediated inhibition of BCL2 and BCLX gene transcription. Notably, downregulation of ICSBP transcripts has been consistently documented in patients with chronic myeloid leukaemia (CML). So, the net effect of BCR–ABL kinase activation is the promotion of cell proliferation and survival through activation of the RAS, SHP2 and PI3K–AKT signalling pathways that lead to increased BCL2 and BCLX expression and inhibition of ICSBP transcription. Small molecules targeting BCR–ABL kinase represent the current mainstay of CML therapy. Pointed arrows indicate direct interactions and/or activations. Blunt-ended arrows indicate inhibitory effects. P, phosphate.
, which in turn binds the scaffolding adaptor GRB2-associated binding protein 2 (GAB2), as well as SOS (a guanine-nucleotide exchanger of RAS), resulting in RAS–MAPK activation (leading to BCL2 gene transcription). On phosphorylation by BCR–ABL, GAB2 recruits SHP2 (also known as PTPN11) and phosphatidylinositol-3 kinase (PI3K), which leads to AKT activation. BCR–ABL also activates other signalling pathways such as signal transducer and activator of transcription 5 (STAT5), leading to BCLX gene (also known as BCL2L1) transcription and the SRC family kinases (SFKs) LYN and HCK. By contrast, BCR–ABL represses interferon consensus sequence binding protein (ICSBP) transcription through an unknown mechanism, which releases the ISCBP-mediated inhibition of BCL2 and BCLX gene transcription. Notably, downregulation of ICSBP transcripts has been consistently documented in patients with chronic myeloid leukaemia (CML). So, the net effect of BCR–ABL kinase activation is the promotion of cell proliferation and survival through activation of the RAS, SHP2 and PI3K–AKT signalling pathways that lead to increased BCL2 and BCLX expression and inhibition of ICSBP transcription. Small molecules targeting BCR–ABL kinase represent the current mainstay of CML therapy. Pointed arrows indicate direct interactions and/or activations. Blunt-ended arrows indicate inhibitory effects. P, phosphate..")
15
Bcr-Abl STAT5 APOPTOSIS PI3 kinase NFB Akt kinase Bax Bak IAPs
Bcl-xL Bad P Bad SMAC P Cyt c Caspase-9 APAF-1, dATP Caspase-9 Caspase-3, -7 APOPTOSIS

16
A) Gruppo 3’-piridilico: aumenta l’attività cellulare del composto
Lead optimization. Development of imatinib from a 2-phenylaminopyrimidine backbone (shown in white). (A) Activity in cellular assays was improved by introduction of a 3' pyridyl group (yellow) at the 3' position of the pyrimidine. (B) Activity against tyrosine kinases was further enhanced by addition of a benzamide group (orange) to the phenyl ring. (C) Attachment of a "flag-methyl" group (green) ortho to the diaminophenyl ring strongly reduced activity against PKC. (D) Addition of an N-methylpiperazine (purple) increased water solubility and oral bioavailability. B) Gruppo amidico: conferisce l’azione anti-TK C) Gruppo metilico: abolisce l’azione anti-PKC D) Gruppo N-metilpiperazinico: aumenta la solubilità e la biodisponibilità per via orale Copyright ©2005 American Society of Hematology. Copyright restrictions may apply.
. (A) Activity in cellular assays was improved by introduction of a 3 pyridyl group (yellow) at the 3 position of the pyrimidine. (B) Activity against tyrosine kinases was further enhanced by addition of a benzamide group (orange) to the phenyl ring. (C) Attachment of a flag-methyl group (green) ortho to the diaminophenyl ring strongly reduced activity against PKC. (D) Addition of an N-methylpiperazine (purple) increased water solubility and oral bioavailability. B) Gruppo amidico: conferisce l’azione anti-TK. C) Gruppo metilico: abolisce l’azione anti-PKC. D) Gruppo N-metilpiperazinico: aumenta la solubilità e la biodisponibilità per via orale. Copyright ©2005 American Society of Hematology. Copyright restrictions may apply.")
17
Figure 1. Schematic representation of the mechanism of action of STI571. The bcr-abl tyrosine kinase is a constitutively active kinase which functions by binding ATP and transferring phosphate from ATP to tyrosine residues on various substrates. This causes the excess proliferation of myeloid cells characteristic of CML. STI571 functions by blocking the binding of ATP to the bcr-abl tyrosine kinase, inhibiting its activity. In the absence of tyrosine kinase activity, substrates required for bcr-abl function cannot be phosphorylated and subsequent cellular events are abrogated.

18
ALTRI BERSAGLI MOLECOLARI DEL GLIVEC

19
Haematological and cytogenetic response in CML: Phase II data
Haematological and cytogenetic response in CML: Phase II data. In all studies, results are expressed as the percentage of responding patients among the patients for whom the diagnosis of the correct phase of chronic myelogenous leukaemia (CML) was confirmed after a central review of the data. A major cytogenetic response combines both complete (0% Ph+ metaphases) and partial (1–35% Ph+) responses. Haematological response was defined as complete haematological response (CHR) in the chronic-phase study, and as either a CHR, a marrow response or a return to chronic phase (RTC) in the advanced-phase studies, all to be confirmed after at least four weeks. In the chronic-phase study, CHR was defined as white blood cells < l-1, platelets < l-1, myelocytes and metamyelocytes <5% in blood, no blasts and promyelocytes in blood, basophils <20% and no extramedullary involvement. In advanced-phase studies, CHR was defined as neutrophils = l-1, platelets = l-1, no blood blasts, marrow blasts <5% and no extramedullary disease. A marrow response was defined by the same criteria as for CHR, but with neutrophils = l-1and platelets = l-1. An RTC was defined as <15% blasts in marrow and blood, <30% blasts and promyelocytes in marrow and blood, <20% basophils in blood and no extramedullary disease. IFN, interferon; Ph+, Philadelphia chromosome positive.
 was confirmed after a central review of the data. A major cytogenetic response combines both complete (0% Ph+ metaphases) and partial (1–35% Ph+) responses. Haematological response was defined as complete haematological response (CHR) in the chronic-phase study, and as either a CHR, a marrow response or a return to chronic phase (RTC) in the advanced-phase studies, all to be confirmed after at least four weeks. In the chronic-phase study, CHR was defined as white blood cells < l-1, platelets < l-1, myelocytes and metamyelocytes <5% in blood, no blasts and promyelocytes in blood, basophils <20% and no extramedullary involvement. In advanced-phase studies, CHR was defined as neutrophils = l-1, platelets = l-1, no blood blasts, marrow blasts <5% and no extramedullary disease. A marrow response was defined by the same criteria as for CHR, but with neutrophils = l-1and platelets = l-1. An RTC was defined as <15% blasts in marrow and blood, <30% blasts and promyelocytes in marrow and blood, <20% basophils in blood and no extramedullary disease. IFN, interferon; Ph+, Philadelphia chromosome positive.")
20
Key points in the discovery and development of Glivec
Key points in the discovery and development of Glivec. The clinical development was particularly rapid, as can be seen by comparison with the typical drug discovery and development times shown in the inset. An NDA for Glivec was submitted just two years and nine months after treatment of the first patient with CML, and FDA approval was given less than three months after application. CML, chronic myelogenous leukaemia; GIST, gastrointestinal stromal tumour; NDA, new drug application; PKC, protein kinase C.

21
Figure 1. Imatinib-induced reduction of CML disease burden
Figure 1. Imatinib-induced reduction of CML disease burden. At diagnosis, chronic-phase CML patients have a disease burden of >1012 leukemia cells. Upon imatinib therapy, >95% of newly diagnosed CML patients re-establish normal blood counts, a process termed complete hematologic response (CHR). The curved arrow indicates progressive levels of response among patients achieving CHR. Non-responders to imatinib therapy ( 5%) are indicated in grey. Most patients (>85%) experience at least a three-log reduction in CML disease burden after imatinib therapy, to a level categorized as minimal residual disease (MRD). Failure to reach this level is viewed as a poor prognostic indicator. Disease levels below 109–1010 leukemic cells generally correspond with complete cytogenetic response, defined as the absence of the t(9;22) in either 20 metaphase cells in a bone marrow aspirate or upon sampling of at least 200 cells in a bone marrow aspirate by fluorescence in situ hybridization. Molecular responses are common, but few patients (<5%) reach the level of PCR negativity (complete molecular response). Thus, almost all responding patients have a residual leukemia burden of >106–107 cells. Measurements of disease burden do not reveal which cell types are susceptible to therapy and which are spared. Thomas O’Hare, Amie S Corbin and Brian J Druker Curr Opin Genet Dev Feb;16(1):92-9. Epub 2005 Dec 15
. The curved arrow indicates progressive levels of response among patients achieving CHR. Non-responders to imatinib therapy ( 5%) are indicated in grey. Most patients (>85%) experience at least a three-log reduction in CML disease burden after imatinib therapy, to a level categorized as minimal residual disease (MRD). Failure to reach this level is viewed as a poor prognostic indicator. Disease levels below 109–1010 leukemic cells generally correspond with complete cytogenetic response, defined as the absence of the t(9;22) in either 20 metaphase cells in a bone marrow aspirate or upon sampling of at least 200 cells in a bone marrow aspirate by fluorescence in situ hybridization. Molecular responses are common, but few patients (<5%) reach the level of PCR negativity (complete molecular response). Thus, almost all responding patients have a residual leukemia burden of >106–107 cells. Measurements of disease burden do not reveal which cell types are susceptible to therapy and which are spared. Thomas O’Hare, Amie S Corbin and Brian J Druker Curr Opin Genet Dev Feb;16(1):92-9. Epub 2005 Dec 15.")
22
Figure 16.25b The Biology of Cancer (© Garland Science 2007)
")
23
Figure 16.30 The Biology of Cancer (© Garland Science 2007)
")
24
Fig. 1. Overview of the different mechanisms of resistance
Fig. 1. Overview of the different mechanisms of resistance. Schematic representation of a cell, the oncogenic kinase and the downstream signaling pathways. P indicates a phosphorylated protein. The concentration of the small molecule inhibitor inside the cell is dependent on its influx and efflux rates. The different sites at which resistance can originate are indicated.

25
Figure 4. Potential mechanisms of disease persistence on imatinib therapy. BCR–ABL is shown in orange; imatinib is in blue. (a) Enhanced drug efflux by ABCG2 (ATPase-binding cassette G2) and/or other transporters (green). (b) BCR–ABL target amplification by increased BCR–ABL transcript levels or gene amplification; insufficient concentration of inhibitor to completely shutdown kinase activity. (c) BCR–ABL kinase domain mutations render persistent cells insensitive to imatinib. (d) Protection within bone marrow microenvironment; stromal cells surround persistent cells. (e) Quiescent, non-cycling cells in deep G0 are impervious to the pro-apoptotic effects of imatinib. (f) BCR–ABL is efficiently targeted, but BCR–ABL kinase activity is dispensable for persistent cell survival
 Enhanced drug efflux by ABCG2 (ATPase-binding cassette G2) and/or other transporters (green). (b) BCR–ABL target amplification by increased BCR–ABL transcript levels or gene amplification; insufficient concentration of inhibitor to completely shutdown kinase activity. (c) BCR–ABL kinase domain mutations render persistent cells insensitive to imatinib. (d) Protection within bone marrow microenvironment; stromal cells surround persistent cells. (e) Quiescent, non-cycling cells in deep G0 are impervious to the pro-apoptotic effects of imatinib. (f) BCR–ABL is efficiently targeted, but BCR–ABL kinase activity is dispensable for persistent cell survival.")
26
Figure 16.26c The Biology of Cancer (© Garland Science 2007)
")
27
Figure 16.27a (part 1 of 2) The Biology of Cancer (© Garland Science 2007)
 The Biology of Cancer (© Garland Science 2007)")
28
Figure 16.27a (part 2 of 2) The Biology of Cancer (© Garland Science 2007)
 The Biology of Cancer (© Garland Science 2007)")
29
Deininger, M. et al. Blood 2005;105:2640-2653
Figure 6. Frequency of BCR-ABL mutations detected in clinical specimens (n = 177) Frequency of BCR-ABL mutations detected in clinical specimens (n = 177). Mutations cluster in 4 distinct regions of the kinase domain. Mutations of the P-loop (amino acids , green) are most common, followed by mutations of T315 (red), which forms a hydrogen bond with imatinib. M351 (turquoise) interacts with the SH2 domain and participates in autoregulation of kinase activity. The fourth cluster (magenta) encompasses the A-loop (amino acids ). Deininger, M. et al. Blood 2005;105: Copyright ©2005 American Society of Hematology. Copyright restrictions may apply.
 Frequency of BCR-ABL mutations detected in clinical specimens (n = 177). Mutations cluster in 4 distinct regions of the kinase domain. Mutations of the P-loop (amino acids , green) are most common, followed by mutations of T315 (red), which forms a hydrogen bond with imatinib. M351 (turquoise) interacts with the SH2 domain and participates in autoregulation of kinase activity. The fourth cluster (magenta) encompasses the A-loop (amino acids ). Deininger, M. et al. Blood 2005;105: Copyright ©2005 American Society of Hematology. Copyright restrictions may apply.")
30
Thr Ile Gln Lys,Val . ABL in complex with imatinib (yellow). The location of mutations are highlighted along with the activation loop (green), nucleotide-binding loop (red), and catalytic loop (orange). strands are numbered and helices are lettered according to the nomenclature used for insulin-receptor tyrosine kinase.[47 and 49] The region including N322 and interacting with the nucleoside-binding loop is shown in cyan.
. The location of mutations are highlighted along with the activation loop (green), nucleotide-binding loop (red), and catalytic loop (orange). strands are numbered and helices are lettered according to the nomenclature used for insulin-receptor tyrosine kinase.[47 and 49] The region including N322 and interacting with the nucleoside-binding loop is shown in cyan.")
31
Figure 16.26b The Biology of Cancer (© Garland Science 2007)
")
32
Figure 2. Early clinical trials results for dasatinib (upper panel) or AMN107 (lower panel) treatment of imatinib-refractory and intolerant CML patients. Results from the two trials are not directly comparable, as a result of shorter follow-up in the AMN107 cohort and slight differences in enrolment criteria. Abbreviations: AP, advanced phase; BC, myeloid blast crisis; CCR, complete cytogenetic response; CHR, complete hematologic response; CP, chronic phase; CR, partial cytogenetic response.

33
a |Surface representations of crystal structures of ABL kinase in complex with imatinib (green), nilotinib (red) and dasatinib (blue). Residues from the nucleotide-binding loop (P-loop) and activation loop (A-loop) are omitted from the surface calculation for clarity. b | Comparison of the different binding modes of three ABL inhibitors: imatinib (left), nilotinib (middle) and dasatinib (right). The positions of the P-loop (red) and A-loop (magenta) vary according to whether the kinase is in an active conformation, in which the P-loop adopts an extended conformation and the N-terminal end of the activation loop adopts a 'DFG-in' conformation (right) or an inactive conformation, in which the P-loop is bent over the inhibitor and the N-terminal end of the activation loop adopts a 'DFG-out' conformation (left and middle). Imatinib and nilotinib block the kinase in an inactive conformation. The green helix is helix C, which often moves between the active and inactive states of kin
 and activation loop (A-loop) are omitted from the surface calculation for clarity. b | Comparison of the different binding modes of three ABL inhibitors: imatinib (left), nilotinib (middle) and dasatinib (right). The positions of the P-loop (red) and A-loop (magenta) vary according to whether the kinase is in an active conformation, in which the P-loop adopts an extended conformation and the N-terminal end of the activation loop adopts a DFG-in conformation (right) or an inactive conformation, in which the P-loop is bent over the inhibitor and the N-terminal end of the activation loop adopts a DFG-out conformation (left and middle). Imatinib and nilotinib block the kinase in an inactive conformation. The green helix is helix C, which often moves between the active and inactive states of kin.")
34
SRC family kinases (SFKs) regulate signal transduction pathways involved in cell growth, differentiation and survival. a | The common central core of SFKs and BCR–ABL is composed of a tyrosine kinase or SRC-homology 1 (SH1) domain, which is preceded by an SH2 domain and an SH3 domain. These three domains are conserved in terms of their sequence homology (42% between ABL and SRC) and arrangement129. By contrast, regions upstream of the SH3 domain and downstream of the kinase domain are variable. The NH2-terminus in the ABL family of kinases is the 'Cap' region, which is present in different splice variants. The homology region in SFK is the N-terminal membrane-localization domain (also referred to as the SH4 domain), which anchors the protein to membranes through myristoyl and/or palmitoyl groups. b | A ribbon representation of the ABL kinase in complex with the ABL and SFK inhibitor PD (Ref. 53). The catalytically active state of ABL kinase closely resembles the active form of SFKs and is often potently inhibited by ATP-competitive molecules originally developed as SFK inhibitors. Dual ABL and SFK inhibitors have proved active against chronic myeloid leukaemia cells exhibiting various forms of imatinib resistance, including BCR–ABL1 overexpression, SFK activation and several BCR–ABL kinase domain mutations.
 domain, which is preceded by an SH2 domain and an SH3 domain. These three domains are conserved in terms of their sequence homology (42% between ABL and SRC) and arrangement129. By contrast, regions upstream of the SH3 domain and downstream of the kinase domain are variable. The NH2-terminus in the ABL family of kinases is the Cap region, which is present in different splice variants. The homology region in SFK is the N-terminal membrane-localization domain (also referred to as the SH4 domain), which anchors the protein to membranes through myristoyl and/or palmitoyl groups. b | A ribbon representation of the ABL kinase in complex with the ABL and SFK inhibitor PD (Ref. 53). The catalytically active state of ABL kinase closely resembles the active form of SFKs and is often potently inhibited by ATP-competitive molecules originally developed as SFK inhibitors. Dual ABL and SFK inhibitors have proved active against chronic myeloid leukaemia cells exhibiting various forms of imatinib resistance, including BCR–ABL1 overexpression, SFK activation and several BCR–ABL kinase domain mutations..")
35
Imatinib behaves as an ATP-competitive inhibitor of the ABL tyrosine kinase with a Ki value of 85 nM130. Imatinib binds to the ATP site and an adjacent small hydrophobic pocket to effectively lock the kinase in an inactive conformation that prevents the transfer of phosphate from ATP to target substrates. By this mechanism imatinib induces cytogenetic responses in the majority of patients with chronic myeloid leukaemia in the chronic phase with a remarkably benign toxicity profile. The development of mutations within the ABL kinase domain has prompted the development of other BCR–ABL inhibitors that have unique modes of action. One such agent, ON012380, blocks the substrate binding site of ABL rather than the ATP pocket, inhibiting this kinase with an IC50 value of 10 nM. Alternatively, other regulatory sites on BCR–ABL are potential targets for small-molecule inhibitors. ABL kinase has a myristoyl binding pocket at the C lobe. Myristoylation stabilizes ABL in its inactive conformation. Although BCR–ABL is not myristoylated, it is expected that compounds that bind to the myristoyl binding pocket can also lock the kinase in its inactive configuration. GNF-2 ((3 [6 [[4-(Trifluoromethoxy)phenyl]amino] 4-pyrimidinyl)benzamide) binds to this myristoyl pocket, resulting in potent ABL kinase inhibition with exceptional enzymatic selectivity.
phenyl]amino] 4-pyrimidinyl)benzamide) binds to this myristoyl pocket, resulting in potent ABL kinase inhibition with exceptional enzymatic selectivity..")
36
The human Aurora kinases (AURKA, AURKB and AURKC) are key mitotic regulators that are required for genomic stability93, 128. Auroras are frequently overexpressed in cancer, which is thought to be important for tumour formation and/or progression. a | The domain structure, number of amino acids, and percentage of sequence homology of human Auroras are depicted. Aurora kinases contain an NH2-terminal regulatory domain (pale orange) and a catalytic kinase domain (grey). Phosphorylation at threonine residues T287/T288 (AURKA), T232 (AURKB) or T195 (AURKC) within the activating T-loops is required for kinase activity. Two sequences, the destruction box (D-Box) and the D-Box-activating domain (DAD/A-Box), promote degradation at the end of mitosis. These features have been confirmed in AURKA and AURKB. However, the structure of AURKC is inferred by sequence alignment and appears to lack the A-Box. AURKA localizes to the duplicated centrosomes and to the spindle poles in mitosis. AURKA overexpression compromises spindle-check-point function and inhibits cytokinesis. AURKB relocates at the onset of anaphase from the chromosomal centrosome to the microtubules that interdigitate at the spindle equator, regulating chromosome condensation by phosphorylating histone H3 (Ref. 93). Aurora kinase inhibitors (AKIs) do not inhibit cell-cycle progression. Rather, cells exposed to AKIs undergo mitosis with normal kinetics and subsequently either re-replicate their genomes, resulting in polyploidy, or undergo apoptosis in a pseudo G1 state. b | This depicts the structure of ABL kinase domain bound to MK-0457, which inhibits all Aurora kinase homologues and the FLT3 kinase that is frequently mutated in acute myeloid leukaemia. Moreover, MK-0457 potently inhibits BCR–ABL T315I kinase, which renders chronic myeloid leukaemia cells insensitive to imatinib, nilotinib and dasatinib. c | The Y-shaped structure of MK-0457 avoids close encounter with leucine in Aurora kinases and isoleucine in BCR–ABL T315 at the gatekeeper position. The four hydrogen bonds within the ABL kinase are depicted (distances in Å).
 and a catalytic kinase domain (grey). Phosphorylation at threonine residues T287/T288 (AURKA), T232 (AURKB) or T195 (AURKC) within the activating T-loops is required for kinase activity. Two sequences, the destruction box (D-Box) and the D-Box-activating domain (DAD/A-Box), promote degradation at the end of mitosis. These features have been confirmed in AURKA and AURKB. However, the structure of AURKC is inferred by sequence alignment and appears to lack the A-Box. AURKA localizes to the duplicated centrosomes and to the spindle poles in mitosis. AURKA overexpression compromises spindle-check-point function and inhibits cytokinesis. AURKB relocates at the onset of anaphase from the chromosomal centrosome to the microtubules that interdigitate at the spindle equator, regulating chromosome condensation by phosphorylating histone H3 (Ref. 93). Aurora kinase inhibitors (AKIs) do not inhibit cell-cycle progression. Rather, cells exposed to AKIs undergo mitosis with normal kinetics and subsequently either re-replicate their genomes, resulting in polyploidy, or undergo apoptosis in a pseudo G1 state. b | This depicts the structure of ABL kinase domain bound to MK-0457, which inhibits all Aurora kinase homologues and the FLT3 kinase that is frequently mutated in acute myeloid leukaemia. Moreover, MK-0457 potently inhibits BCR–ABL T315I kinase, which renders chronic myeloid leukaemia cells insensitive to imatinib, nilotinib and dasatinib. c | The Y-shaped structure of MK-0457 avoids close encounter with leucine in Aurora kinases and isoleucine in BCR–ABL T315 at the gatekeeper position. The four hydrogen bonds within the ABL kinase are depicted (distances in Å)..")
37
ALTRI BERSAGLI MOLECOLARI DEL GLIVEC

38
Gastrointestinal stromal tumors
ULTERIORI APPLICAZIONI TERAPEUTICHE DEL GLIVEC GIST Gastrointestinal stromal tumors

39
Figure 16.29 The Biology of Cancer (© Garland Science 2007)
")
40
Chronic myelomonocytic leukemia
ULTERIORI APPLICAZIONI TERAPEUTICHE DEL GLIVEC CMML Chronic myelomonocytic leukemia

41
Small cell lung carcinomas
ULTERIORI APPLICAZIONI TERAPEUTICHE DEL GLIVEC Glioblastomi SCLC Small cell lung carcinomas

42
EFFETTI DEL GLIVEC SUL MICROAMBIENTE TUMORALE
Inibizione degli effetti angiogenici del PDGF sulle cellule dello stroma sulle cellule endoteliali

43
A: Effects of imatinib on sprout formation induced by FCS in the rat aortic square assay. Pieces (1 x 1 mm) of rat aorta were grown in a fibrin gel in the presence of 10% fetal calf serum and increasing concentrations of imatinib ( I~M) or vehicle (control) (x-axis). Samples were then incubated for six days. Aminocaproic acid (300 I~g/ml) was added on the first three days to prevent degradation of the gel. The media was changed on the fourth day, with fresh additions of all required factors and 50 I~g/ml aminocaproic acid. The area of sprouts formed was measured from the image viewed under an inverse microscope using a KS-400 imaging Imatinib induced concentration-dependent inhibition of the formation of sprouts. Two experiments were performed that showed the same results. Data presented in the graph are pooled from the two experiments. Results are presented as mean 4- SEM, n = 7-8. *P < significance compared to control (Tukey test). Effects of imatinib on sprout formation induced by FCS in the rat aortic square assay. Pieces (1 x 1 mm) of rat aorta were grown in a fibrin B: Porous chambers containing VEGF (2 I~g/ml), bFGF (0.3 I~g/ml) or PDGF (3 I~g/ml) in 0.8% w/v agar containing heparin (20 U/ml) were implanted subcutaneously in the flank of Tiflbm : MAG mice. The growth factors induce the growth of vascularized tissue around the chamber. This response is quantified by measuring the weight and blood content of the tissue. Mice were treated with imatinib (50 mg/kg p.o. twice daily) starting one day before implantation of the chambers and the animals were sacrificed for measurement of the vascularized tissues after five days of treatment. Values are mean 4- SEM, n = 11, *P < 0.05 (rank sum test) significant inhibition compared to the response in the group receiving growth factor alone.
 or vehicle (control) (x-axis). Samples were then. incubated for six days. Aminocaproic acid (300 I~g/ml) was added on the first three days to prevent degradation of the gel. The media was changed on. the fourth day, with fresh additions of all required factors and 50 I~g/ml aminocaproic acid. The area of sprouts formed was measured from the image. viewed under an inverse microscope using a KS-400 imaging Imatinib induced concentration-dependent inhibition of the formation of. sprouts. Two experiments were performed that showed the same results. Data presented in the graph are pooled from the two experiments. Results are. presented as mean 4- SEM, n = 7-8. *P < significance compared to control (Tukey test). Effects of imatinib on sprout formation induced by FCS in the rat aortic square assay. Pieces (1 x 1 mm) of rat aorta were grown in a fibrin. B: Porous chambers containing VEGF (2 I~g/ml), bFGF (0.3 I~g/ml) or PDGF (3 I~g/ml) in 0.8% w/v agar containing heparin (20 U/ml) were implanted subcutaneously in the flank of Tiflbm : MAG mice. The growth factors induce the growth of vascularized tissue around the chamber. This response is quantified by measuring the weight and blood content of the tissue. Mice were treated with imatinib (50 mg/kg p.o. twice daily) starting one day before implantation of the chambers and the animals were sacrificed for measurement of the vascularized tissues after five days of treatment. Values are mean 4- SEM, n = 11, *P < 0.05 (rank sum test) significant inhibition compared to the response in the group receiving growth factor alone.")
44
EFFETTI TOSSICI DEL GLIVEC
Nausea; vomito Ritenzione di liquidi Crampi muscolari e dolori articolari Mielosoppressione di grado 2-3 Cardiotossicità

45
a | Constitutive signalling in chronic myeloid leukaemia (CML) progenitor cell, through the cytoplasmic BCR–ABL tyrosine kinase, leads to activation of Ras–ERK (extracellular signal-regulated kinase), phosphatidylinositol 3-kinase (PI3K)–Akt and signal transducer and activator of transcription 5 (STAT5) pathways. Several of these pathways converge on anti-apoptotic mechanisms: the Ras–ERK pathway stimulates the expression of BCL2, STAT5 activates BCL-X, and Akt inhibits BCL2-antagonist of cell death (BAD) and forkhead box O3A (FOXO3A). Imatinib blocks all BCR–ABL-dependent phosphorylation and signalling events, leading to reversal of pro-survival effects and activation of apoptosis. b | In cardiomyocytes ABL (localized to plasma membrane or endoplasmic reticulum, ER) seems to maintain ER homeostasis by mechanisms that are not yet clear. The ABL kinase inhibitor, imatinib, induces ER stress, leading to activation of the PKR-like ER kinase (PERK) and IRE1 pathways, and to overexpression of protein kinase C (PKC ). PERK phosphorylates the eukaryotic translation initiation factor 2 (EIF2 ) as part of a protective response, and on sustained ER stress IRE1 activates Jun N-terminal kinases (JNKs), leading to phosphorylation of and release of BAX followed by mitochondrial depolarization, ATP depletion, cytochrome c (Cyt c) release, and features of necrotic and apoptotic cell death , protein; ASK1, apoptosis signal-regulating kinase 1; BAX, BCL2 associated X protein; GAB2, GRB2-associated binding protein 1; GRB2, growth factor receptor-bound protein 2; MEK, mitogen-activated ERK kinase; PIP3, phosphatidylinositol trisphosphate; SOS, son of sevenless.
 and forkhead box O3A (FOXO3A). Imatinib blocks all BCR–ABL-dependent phosphorylation and signalling events, leading to reversal of pro-survival effects and activation of apoptosis. b | In cardiomyocytes ABL (localized to plasma membrane or endoplasmic reticulum, ER) seems to maintain ER homeostasis by mechanisms that are not yet clear. The ABL kinase inhibitor, imatinib, induces ER stress, leading to activation of the PKR-like ER kinase (PERK) and IRE1 pathways, and to overexpression of protein kinase C (PKC ). PERK phosphorylates the eukaryotic translation initiation factor 2 (EIF2 ) as part of a protective response, and on sustained ER stress IRE1 activates Jun N-terminal kinases (JNKs), leading to phosphorylation of and release of BAX followed by mitochondrial depolarization, ATP depletion, cytochrome c (Cyt c) release, and features of necrotic and apoptotic cell death , protein; ASK1, apoptosis signal-regulating kinase 1; BAX, BCL2 associated X protein; GAB2, GRB2-associated binding protein 1; GRB2, growth factor receptor-bound protein 2; MEK, mitogen-activated ERK kinase; PIP3, phosphatidylinositol trisphosphate; SOS, son of sevenless..")
Presentazioni simili











>")
>")








