Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Accumuli Intracellulari
Gli accumuli intracellulari avvengono in risposta ad alterazioni metaboliche e possno essere di vario tipo: Costituenti Normali endogene: Accumuli di acidi grassi, proteine, carboidrati, acqua Costituenti anomali endogene: proteine con conformazione alterata (aggregati) Costituenti Anomali esogeni Minerali o prodtti di agenti infettivi (tossine) Pigmenti: sostanze colorate esogene (carbone, silice) o endogene (melanina)
 Costituenti Anomali esogeni. Minerali o prodtti di agenti infettivi (tossine) Pigmenti: sostanze colorate esogene (carbone, silice) o endogene (melanina)")
2
Accumuli intracellulari di sostanze endogene
Accumulo di sostanze normali endogene Accumulo di sostanze endogene può conseguire a difetti genetici che causano problemi di metabolismo, di trasporto o di deposito

3
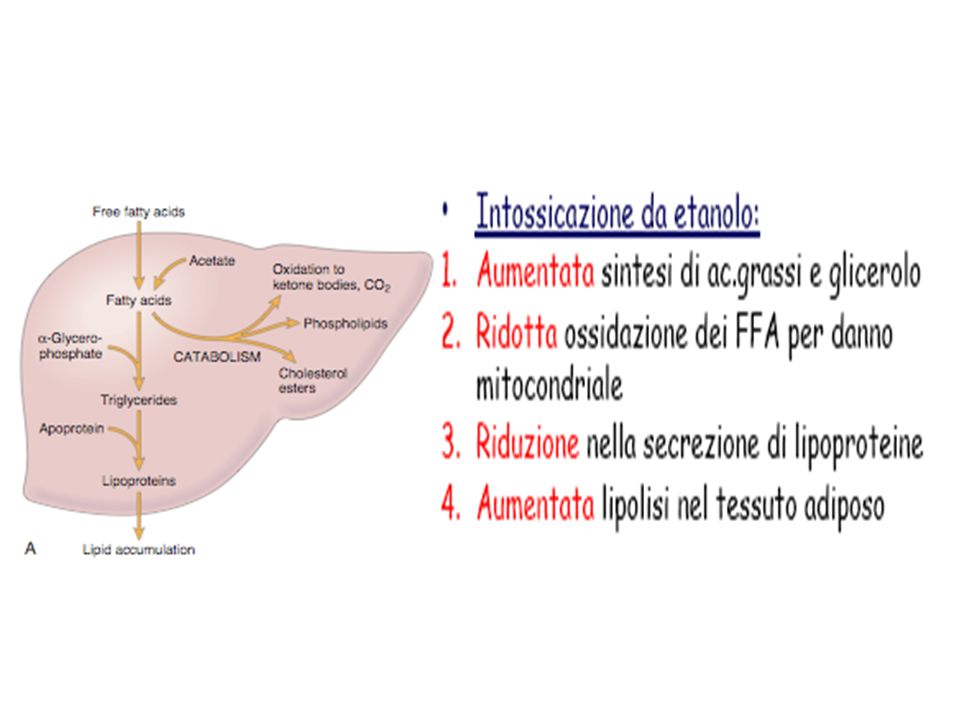
Accumulo di Lipidi e acidi grassi
Accumulo di sostanza endogena normale per ridotto metabolismo: Accumili di lipidi e acidi grassi (steatosi epatica, degenerazione grassa per accumulo di trigliceridi) Accumulo di Lipidi e acidi grassi Dal tessuto adiposo Steatosi o degenerazione grassa del fegato: Normalmente, gli acidi grassi provengono dal tessuto adiposo o sono sintetizzati a partire dall’acetato e vengono: esterificati in trigliceridi, associati ad apoproteine e secreti come lipoproteine convertiti a colesterolo o fosfolipidi ossidati a corpi chetonici
 Accumulo di Lipidi e acidi grassi. Dal tessuto adiposo. Steatosi o degenerazione grassa del fegato: Normalmente, gli acidi grassi provengono dal tessuto adiposo o sono sintetizzati a partire dall’acetato e vengono: esterificati in trigliceridi, associati ad apoproteine e secreti come lipoproteine. convertiti a colesterolo o fosfolipidi. ossidati a corpi chetonici.")
4
L’accumulo di lipidi può essere la conseguenza di difetti delle normali vie catabiliche o metaboliche delgi acidi grassi: esempi: L’anossia inibisce l’ ossidazione in corpi chetonici Epatotossine alterano le funzioni mitocondriali e quindi l’ossidazione degli acidi grassi Il digiuno aumento la mobilitazione dei depositi periferici

6
La Steatosi è provocata generalmente da cause esogene (agenti vari o a carenza di fattori vari)
Tipi di steatosi: da etanolo da sovraccarico da aumento di sintesi di acidi grassi da carenza di proteine da carenza di fosfolipidi per inibizione della sintesi proteica da tossine da CCl 4 da blocco della secrezione

7
Steatosi geneticamente determinata
M. di Wolman: colpisce gli istiociti e le cellule parenchimali epatiche, deficienza genetica: lipasi acida Deficienza ereditaria della b-lipoproteina: colpisce le cellule della mucosa intestinale, deficienza genetica : apolipoproteina che dovrebbe combinarsi con i trigliceridi e portarli nel circolo linfatico

8
ACCUMULO – Grasso nel Fegato - Alcolismo

9
Infiammazione e necrosi: macrofagi che accumulano colesterolo perchè fagocitano membrane di cellule danneggiate in luoghi in cui c’è danno o infiammazione in atto

10
Causa: poligenica, stile di vita
Aterosclerosi Causa: poligenica, stile di vita Ossidazione della componenete lipidica dell’LDL (lipoproteina che lega e trasporta il Colesterolo) Sviluppo di placche fibrose (con nucleo centrale ricco di colesterolo) nello strato di cellule endoteliali più interno dei vasi sanguigni (intima): ischemia del tessuto cardiaco e cerebrale Introdurre l’importanza dei lipidi
 Sviluppo di placche fibrose (con nucleo centrale ricco di colesterolo) nello strato di cellule endoteliali più interno dei vasi sanguigni (intima): ischemia del tessuto cardiaco e cerebrale. Introdurre l’importanza dei lipidi.")
11
Accumulo si sostanza anomala esogena verso cui non si hanno gli enzimi digestivi e pertanto si accumula (carbone, silice)
")
12
S.R.E Sistema reticolo endoteliale
Cellule del sistema immunitario: Cellule reticolari e macrofagi collocati negli organi linfoidi primari e secondari: Midollo osseo, Milza, linfonodi, fegato Il ferro di riserva è presente sotto due forme: come ferritina e come emosiderina. La ferritina è un complesso idrosolubile costituito da un nucleo centrale di idrossido di ferro e da una proteina, la apoferritina che lega il ferro La presenza del ferro stimola la formazione di apoferritina, ma se la quantità di ferro nelle cellule è molto elevata la quantità di apoferritina che si forma non è sufficiente per legare tutto il ferro e questo si deposita in forma di emosiderina. La ferritina è largamente diffusa nell'organismo trovandosi, oltre che nelle cellule del S.R.E., anche nelle cellule di numerosi tessuti (reticolociti, placenta, testicoli, rene, miocardio, muscoli scheletrici, pancreas ecc.) e nelle cellule della mucosa intestinale dove riveste particolare importanza nell'assorbimento e nel metabolismo del ferro.
 e nelle cellule della mucosa intestinale dove riveste particolare importanza nell assorbimento e nel metabolismo del ferro.")
13
Livido

14
Melanina: ossidazione della tirosina e della diidrossifenilalanina nei melanociti

15
Polvere di carbone: captato dai macrofagi degli alveoli e trasportato ai linfonodi regionali. Gli aggregati di carboni possono indurre una reazione fibroblastica o enfisema

16
Formazione di fosfati di calcio in forma di apatite simile all’idrossiapatite dell’osso
Accumuli Intracellulari: Inizia nei mitocndri delle cellule necrotiche Accumuli extracellulari: inizia dai fosfolipidi delle vescicole che si formano in prossimià della membrana di cellule danneggiate o invecchiate

18
Accumuli di proteine Cause di accumuli intracellulari di proteine (vacuoli, gocciole eusinofile) Gocciole di riassorbimento a livello dei tubuli prossimali del rene: osservabile nelle patologie renali che causano la perdita di proteine nelle urine, indicendo così un aumento del riassorbimento proteico Sintesi eccessiiva di proteine di secrezione, avviene nelle plasmacellule (linfociti B maturi che severnano Anticorpi) Difetti del ripiegamento “folding” proteico che possono indurre l’accumolo di aggregati proteici
 Difetti del ripiegamento folding proteico che possono indurre l’accumolo di aggregati proteici.")
19
Folding proteico Una catena polipeptidica può assumere diverse possibili conformazioni che vanno dallo stato “unfolded” (random coil), senza struttura definita, a quello ripiegato (folded) o nativo. A bassa T la G dello stato nativo è minore della G dello stato unfolded e quindi la proteina assume conformatione nativa Le diverse possibili conformazioni assunte corrispondono a quelle “termodinamicamente” più stabili e cioè a quelle che hanno un energia libera (G) minore ad una data temperatura (T) o concentrazione Numero interazioni native Stato unfold Energia libera Stato nativo Numero contatti tra aa
, senza struttura definita, a quello ripiegato (folded) o nativo. A bassa T la G dello stato nativo è minore della G dello stato unfolded e quindi la proteina assume conformatione nativa. Le diverse possibili conformazioni assunte corrispondono a quelle. termodinamicamente più stabili e cioè a quelle che hanno un energia libera (G) minore ad una data temperatura (T) o concentrazione. Numero interazioni native. Stato unfold. Energia libera. Stato nativo. Numero contatti tra aa.")
20
Numero interazioni native
Stato unfold Energia libera Stato nativo Numero contatti tra aa I possibili legami deboli all’interno della una catena polipeptidica (tra gli amminoacidi) e le forze idrofobiche sono determinanti per I possibili ripiegamenti della catena polipeptidica e pertanto la possibile conformazione nativa è intrinsicamente contenuta nella struttura primaria di una proteina (sequenza di aa) ed infatti è possibile far avvenire il folding nativo in vitro. Però i tempi o temperatue richiesti nella maggioranza dei casi non sarebbero compatibili con la fisiologia cellulare. Pertanto il folding proteico viene mediato dai CHAPERONI MOLECOLARI che sono delle proteine capaci di legare i polipeptidi nello stato “non nativo” cioè “unfolded” e ne consentono il folding corretto
 e le forze idrofobiche sono determinanti per I possibili ripiegamenti della catena polipeptidica e pertanto la possibile conformazione nativa è intrinsicamente contenuta nella struttura primaria di una proteina (sequenza di aa) ed infatti è possibile far avvenire il folding nativo in vitro. Però i tempi o temperatue richiesti nella maggioranza dei casi non sarebbero compatibili con la fisiologia cellulare. Pertanto il folding proteico viene mediato dai CHAPERONI MOLECOLARI che sono delle proteine capaci di legare i polipeptidi nello stato non nativo cioè unfolded e ne consentono il folding corretto.")
21
Chaperoni molecolari In assenza di chaperoni molecolari le proteine con più di 100 aa (90% delle proteine cellulari) tendono a collapsare in strutture globulari compatte (aggregati amorfi). In alternativa, si possono formare strutture fibrillari che danno luogo ai depositi di amiloide, coinvolti in numerose malattie degenerative
 tendono a collapsare in strutture globulari compatte (aggregati amorfi). In alternativa, si possono formare strutture fibrillari che danno luogo ai depositi di amiloide, coinvolti in numerose malattie degenerative.")
22
I chaperoni molecolari sono di due tipi:
Legano le catene polipeptiidiche nascenti e ne consentono il folding 2. Legano le proteine”unfolded” dopo che la traduzione è stata completata Tipo 1 chaperon di piccole dimensioni, KDa che sono indicati normalmente come chaperoni. Tipo 2 chaperoni di grandi dimensioni, 8000 KDa, che sono indicati come chaperonine.

23
Chaperoni di tipo 1: heat shock proyein (HSP)
Gli chaperoni prevengono le interazioni aspeifiche legandosi ai residui idrofobici, impedendone l’intereazione con altri residui idrofobici di proteine vicine (o l’’interno della stessa proteina) e pertanto impediscono la formazione degli aggregati insolubili Le catene nascenti sui polisomi sono prone ad interazioni non specifiche che inducno aggregazione Questo processo richiede energia che viene fornita dalla scissione dell’ ATP in ADP da parte degli chaperoni che hanno attività ATPasica
 e pertanto impediscono la formazione degli aggregati insolubili. Le catene nascenti sui polisomi sono prone ad interazioni non specifiche che inducno aggregazione. Questo processo richiede energia che viene fornita dalla scissione dell’ ATP in ADP da parte degli chaperoni che hanno attività ATPasica.")
24
Chaperoni molecolari di tipo 2: chaperonine di classe II
Presenti in archaea ed eucarioti (corrispondenti ai chparoni GroEL-Gro-ES-dei batteri) Sono grossi complessi proteici strutturati in due anelli formati da 8-10 subunità. Ciascuna unità ha un ATP-binding domain, un dominio intermedio ed uno apicale che lega il peptide. Un estrusione della parte apicale forma il coperchio di chiusura In assenza di ATP le subunità sono in conformazione aperta, in presenza di ATP avviene il legame della proteina unfolded e si ha un cambio conformazionale L’idrolisi dell’ATP comporta il folding ed il rilascio della proteina nella cavità centrale Strutture dei cristalli nella forma aperta e chiusa
 Sono grossi complessi proteici strutturati in due anelli formati da 8-10 subunità. Ciascuna unità ha un ATP-binding domain, un dominio intermedio ed uno apicale che lega il peptide. Un estrusione della parte apicale forma il coperchio di chiusura. In assenza di ATP le subunità sono in conformazione aperta, in presenza di ATP avviene il legame della proteina unfolded e si ha un cambio conformazionale. L’idrolisi dell’ATP comporta il folding ed il rilascio della proteina nella cavità centrale. Strutture dei cristalli nella forma aperta e chiusa.")
25
Le chaperonine funzionano in coperazione con Hsp70 nel procura il folding delle proteine
Chaperon: HSP70/Hsp40 Chaperonina

26
Esempi di patologie dovute alla formazione di aggregati proteici:
Alzheimer’s disease APP β-peptide Polyglutamine expansion diseases (CAG) Various polyQ proteins Creutzfeldt–Jakob’s disease Prion protein Cirrosi epatica a-antitripsina Mutazioni nei chaperoni molecolari: Hereditary spastic paraplegia Mitochondrial Hsp60 Mancanza di accumulo di proteina nella sua naturale locazione Cystic fibrosis CFTR Familial hypercholesterolemia LDL receptor
 Various polyQ proteins. Creutzfeldt–Jakob’s disease Prion protein. Cirrosi epatica a-antitripsina. Mutazioni nei chaperoni molecolari: Hereditary spastic paraplegia Mitochondrial Hsp60. Mancanza di accumulo di proteina nella sua naturale locazione. Cystic fibrosis CFTR. Familial hypercholesterolemia LDL receptor.")
27
In assenza di folding corretto,
alcune proteine sono riconosciute come substrato per gli enzimi del sistema di degradazione “uniquitina-Proteosoma

28
Più molecole di ubiquitina vengono trasferite dagli enzimi E1
Più molecole di ubiquitina vengono trasferite dagli enzimi E1. E2 and E3 (Ubiquitin-ligasi) alla proteina target Quando taggate dall’ubiquitina, le proteine entrano nel proteosoma, un multicomplesso proteico a forma di cilndro, all’interno del quale avviene la degradazione delle proteine ubiquitinate e lo stacco delle ubquitine che ritornano in ciclo
 alla proteina target. Quando taggate dall’ubiquitina, le proteine entrano nel proteosoma, un multicomplesso proteico a forma di cilndro, all’interno del quale avviene la degradazione delle proteine ubiquitinate e lo stacco delle ubquitine che ritornano in ciclo.")
29
Nella Fibrosi Cistica, La mutazione più frequentedella CFTR consiste nella delezione della fenilalanina (DF508) che induce un diffetto di folding , ritensione nel reticolo endoplasmatico (ER) e la conseguente degradazione della proteina dal parte del proteasoma Pertanto l’uso di inibitori farmacologici del proteasoma può allievare la patologia dovuta alla mutazione DF508 in quanto provocherebbe un aumento della CFTR che raggiunge la mambrana citoplasmatica

30
GENE MUTATO E SUA FUNZIONE
Il gene mutato è il CFTR e codifica una proteina di membrana la cui funzione è quella di trasportare il cloro attraverso le membrane delle cellule epiteliali dei: Alveoli bronchiali pancreas, intestino, ghiandole salivari, ghiandole sudoripare vasi deferenti.

31
CFTR è un “ligand-dependent” Cl chanel l’ ATP intracellulare funziona da ligando
L’apertura del canale dipende dal binding dell’ATP e dall’attivazione del dominio di regolazione che viene indotta per fosforilazione R-domain: Dominio di Regolazione Contiene siti di fosforilazione per la cAMP-dependent PKA e per la PKC, due chinasi che sono centrali nella fisiologia cellulare di tutti I tipi cellulari (trasduzione del segnale), in quanto regolano pathway cellulari che vanno dall’espressione genica (fosforilazione di fattori di trascizione) all’attivazione di domini proteici enzimatici. La PKA è attivata dall’AMP ciclico, mentre la PKC è attivata dal diacilglicerolo (DAG) o dai ioni Ca++
, in quanto regolano pathway cellulari che vanno dall’espressione genica (fosforilazione di fattori di trascizione) all’attivazione di domini proteici enzimatici. La PKA è attivata dall’AMP ciclico, mentre la PKC è attivata dal diacilglicerolo (DAG) o dai ioni Ca++")
32
Fibrosi cistica La fibrosi cistica(FC) o mucoviscidosi è una malattia congenita, cronica che coinvolge numerosi organi ed apparati, come: l’apparato respiratorio, il pancreas, il fegato, l’intestino l’apparato riproduttivo maschile (vasi deferenti) La malattia può manifestarsi precocemente, con gravità diversa, più raramente la malattia può evidenziarsi nell’età adolescenziale o adulta con quadri clinici meno gravi.
 La malattia può manifestarsi precocemente, con gravità diversa, più raramente la malattia può evidenziarsi nell’età adolescenziale o adulta con quadri clinici meno gravi.")
33
Sudore salato : dovuto ad elevato contenuto di Cloro nel sudore
Test diagnostico principale consiste nello stimolare le ghiandole del sudore della pelle dell’avambraccio, raccogliere il sudore e dosarvi il cloro: si fa diagnosi di fibrosi cistica quando il cloro nel sudore è elevato, perciò maggiore di 60 mg/L. Ghiandola sudoripara normale Ghiandola sudoripara in CF Il riassorbimento del Cl- e del Na+ non avviene nelle ghiandole dei pazienti CF La ghiandola secerne sudore, (H2O, Cl-, Na+, K+, urea, colesterolo e acidi grassi); il riassorbimento dei sali inizia ad opera del canale del Na+, a cui segue il riassorbito del Cl- Nelle ghiandole sudoripare ci sono altri canali per il Cloro, quindi nei pazienti CF la secrezione avviene, ma il riassorbimento del cloro e sodio è ridotto
; il riassorbimento dei sali inizia ad opera del canale del Na+, a cui segue il riassorbito del Cl- Nelle ghiandole sudoripare ci sono altri canali per il Cloro, quindi nei pazienti CF la secrezione avviene, ma il riassorbimento del cloro e sodio è ridotto.")
36
Atrofia corticale che porta alla perdita di attività cerebrale
Malattia di Alzheimer Atrofia corticale che porta alla perdita di attività cerebrale Alterazione strutturale della proteina precursore dell’ amiloide APP Cause: 1% mutazione genetica 99% sporadica, Invecchiamento, stile di vita MRI scanning Imaging a risonanza magnetica

37
Esempi di mutazioni missense in patologie umane
Nell’Alzheimer familiare (causa genetica) mutazioni missense che sostituiscono gli aa in rosso, alterano il taglio proteolitico della proteina precusrore amieloide (APP) da parte delle b- e g-secretasi e si generano peptidid B-amiloidi con aumentata capacità a formare aggragati
 mutazioni missense che sostituiscono gli aa in rosso, alterano il taglio proteolitico della proteina precusrore amieloide (APP) da parte delle b- e g-secretasi e si generano peptidid B-amiloidi con aumentata capacità a formare aggragati.")
Presentazioni simili



















>")


