Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
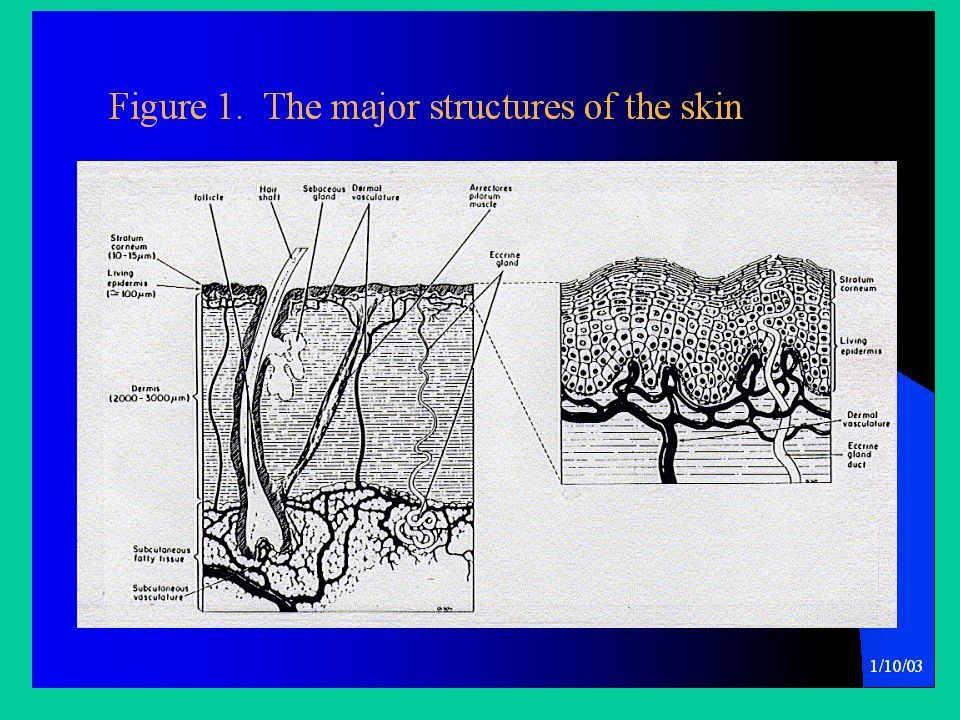
ASSORBIMENTO TRANSCUTANEO
La cute integra è un’ottima barriera verso molti agenti chimici. Tuttavia sostanze sia gassose che liquide possono essere assorbite attraverso la cute in quantità sufficiente a dare tossicità sistemica. L’assorbimento avviene principalmente per diffusione Alcuni farmaci sono somministrati per via transdermica (cerotti, gel); si ottengono livelli plasmatici costanti per un lungo periodo (24 ore o più). Questa via consente anche di evitare il metabolismo di I passaggio. Importante in tossicologia. Ad es., diversi insetticidi possono essere ben assorbiti ed hanno in passato causato morti in lavoratori agricoli. La superficie assorbente è 1,5-2 m2 nell’adulto
; si ottengono livelli plasmatici costanti per un lungo periodo (24 ore o più). Questa via consente anche di evitare il metabolismo di I passaggio. Importante in tossicologia. Ad es., diversi insetticidi possono essere ben assorbiti ed hanno in passato causato morti in lavoratori agricoli. La superficie assorbente è 1,5-2 m2 nell’adulto.")
2
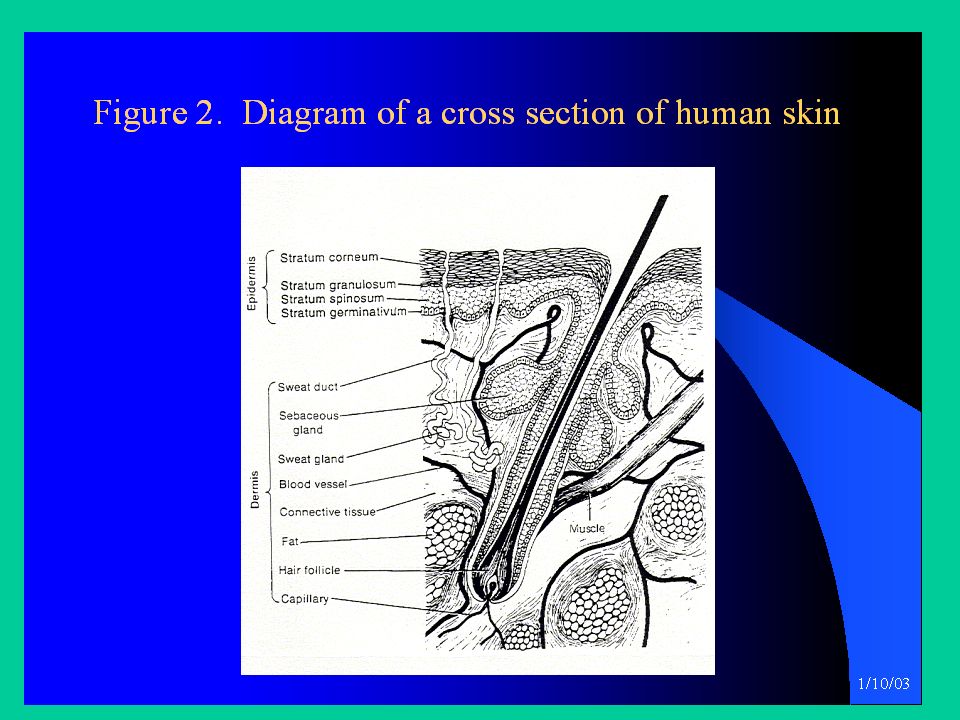
Le sostanze che vengono assorbite per via transcutanea devono passare attraverso 7 strati cellulari prima di raggiungere i capillari e i vasi linfatici del derma. La barriera principale all’assorbimento, che limita la velocità di assorbimento, è rappresentata dallo strato più esterno dell’epidermide, lo strato corneo. Le cellule di questo strato durante il loro ciclo, perdono il nucleo, si disidratano e la loro matrice cellulare polimerizza, dando luogo a cellule ‘secche’ e piene di cheratina. Le pareti cellulari si ispessiscono per l’inclusione di materiali chimicamente inerti. Le cellule cheratinizzate formano uno strato altamente ‘impaccato’.

6
Il risultato è una drastica diminuzione della diffusibilità delle sostanze attraverso il tessuto.
L’assorbimento cutaneo è in genere più lento e meno esteso e più selettivo rispetto ad altre vie (orale, polmonare). Un fattore importante che condiziona la permeabilità attraverso lo strato corneo è la sua composizione in lipidi; lo strato corneo è infatti ricco di lipidi relativamente polari, quali ceramidi, colesterolo ed esteri del colesterolo, mentre sono assenti i trigliceridi. In virtù di questa composizione, lo strato corneo può essere attraversato da molecole lipofile aventi un certo grado di idrofilia. Molecole estremamente lipofile, quali TCDD, hanno un assorbimento minimo, così come molecole idrofile.
. Un fattore importante che condiziona la permeabilità attraverso lo strato corneo è la sua composizione in lipidi; lo strato corneo è infatti ricco di lipidi relativamente polari, quali ceramidi, colesterolo ed esteri del colesterolo, mentre sono assenti i trigliceridi. In virtù di questa composizione, lo strato corneo può essere attraversato da molecole lipofile aventi un certo grado di idrofilia. Molecole estremamente lipofile, quali TCDD, hanno un assorbimento minimo, così come molecole idrofile.")
8
Alcune sostanze (es. steroidi) possono essere assorbite attraverso i follicoli piliferi, che rappresentano però solo lo 0,2% della superficie corporea totale. Lo strato corneo può funzionare da “deposito”. Le sostanze assorbite si depositano prima sulla parte superiore del corneo; in una seconda fase, allorché si raggiunge una condizione di sovrasaturazione, permeano gli strati più interni. Ad es., gli steroidi restano sulla cute per 7-10 giorni dopo una singola applicazione. E’ stato osservato che l’emivita plasmatica di idrocortisone e clobetasone è di diversi giorni, dopo una singola applicazione cutanea.

9
sostanze polari sostanze lipofile

10
La velocità di assorbimento transcutaneo aumenta con la temperatura.
Tmax (ore) - 18° 12,2 2° 10,4 20° 8,5 51° 5,6 Tempo necessario per l’effetto tossico massimo dopo applicazione del gas nervino XV allo stato liquido
 - 18° 12,2. 2° 10,4. 20° 8,5. 51° 5,6. Tempo necessario per l’effetto tossico massimo dopo applicazione del gas nervino XV allo stato liquido.")
11
L’assorbimento transcutaneo varia in funzione della permeabilità dello strato corneo
Regione esposta: la permeabilità è maggior e nelle zone dove lo strato corneo è più sottile: regione scrotale > cuoio capelluto > avambraccio > regione plantare Integrità dello strato corneo: acidi e basi forti, gas mostarda e altri agenti lesionano lo strato corneo e ne aumentano la permeabilità. Solventi come il DMSO aumentano la permeabilità perché rimuovono i lipidi e agiscono da idratanti. Ustioni e malattie cutanee aumentano l’assorbimento. Idratazione: aumenta la permeabilità. Nei bambini la permeabilità è maggiore perché lo strato corneo è più idratato e più sottile.

12
L’assorbimento transcutaneo di un tossico liquido diminuisce all’aumentare della volatilità
Volatilità (mg/m3 a 20° C) DL50 (mg liquido) VX 10 Soman 2.650 50 – 300 Sarin 6.100 100 – 500
 DL50. (mg liquido) VX. 10. Soman – 300. Sarin – 500.")
13
Differenze tra specie. Importanti per la tossicità selettiva degli insetticidi. Ad es., il DDT ha una DL50 simile negli insetti e nei mammiferi se iniettato. Dopo applicazione esterna, invece, il DDT è molto meno tossico nei mammiferi rispetto agli insetti perché: l’esoscheletro di chitina degli insetti è molto più permeabile; il rapporto superficie corporea/massa corporea è molto maggior negli insetti.

14
Vie d’assorbimento parenterali sistemiche
Via intravascolare (endovenosa [e.v.], intrarteriosa): assorbimento rapido e completo. Utile in caso di necessità di azione immediata e per farmaci poco assorbiti per altre vie o irritanti per via intramuscolare. Il farmaco può essere somministrato in unica iniezione (bolo) o infuso a velocità regolata.
: assorbimento rapido e completo. Utile in caso di necessità di azione immediata e per farmaci poco assorbiti per altre vie o irritanti per via intramuscolare. Il farmaco può essere somministrato in unica iniezione (bolo) o infuso a velocità regolata.")
15
Rischi e precauzioni: somministrazione troppo rapida effetti tossici improvvisi iniettare lentamente. materiali o soluzioni non sterili infezioni infusione di sostanze oleose, di aria, precipitati nella soluzione embolismi soluzioni ipertoniche aggregazione globuli rossi soluzioni ipotoniche emolisi

16
Via intramuscolare (i. m
Via intramuscolare (i.m.): utile per assorbimento rapido e per molecole poco assorbite o degradate a livello GI. Può provocare dolore, ascessi o necrosi al sito di iniezione, se la soluzione iniettata è irritante. Via sottocutanea (s.c.), via intradermica. assorbimento più lento rispetto a i.m. utile per mantenere livelli plasmatici più a lungo (es. insulina). Formulazioni ‘deposito’, che rilasciano il farmaco lentamente, possono essere impiantate sottocute (es. ormoni steroidei). Via intraperitoneale (i.p.): solo sperimentale (animali). La soluzione viene iniettata nella cavità peritoneale: assorbimento rapido e completo.
: utile per assorbimento rapido e per molecole poco assorbite o degradate a livello GI. Può provocare dolore, ascessi o necrosi al sito di iniezione, se la soluzione iniettata è irritante. Via sottocutanea (s.c.), via intradermica. assorbimento più lento rispetto a i.m. utile per mantenere livelli plasmatici più a lungo (es. insulina). Formulazioni ‘deposito’, che rilasciano il farmaco lentamente, possono essere impiantate sottocute (es. ormoni steroidei). Via intraperitoneale (i.p.): solo sperimentale (animali). La soluzione viene iniettata nella cavità peritoneale: assorbimento rapido e completo.")
17
Dose e livelli plasmatici
All’aumentare della dose somministrata, aumenta in modo proporzionale la quantità di farmaco assorbito aumento della concentrazione plasmatica. Dato che l’eliminazione segue una cinetica di I ordine, all’aumentare della concentrazione plasmatica aumenta anche la velocità di eliminazione.

18
200 mg kel: 1 h-1 100 mg

19
200 mg; kel 0,5 h-1 100 mg; kel 1 h-1

20
Quasi sempre, la concentrazione plasmatica ad un certo tempo è direttamente proporzionale alla dose somministrata. Per pochi farmaci, i meccanismi di eliminazione sono saturabili. In questi casi, la concentrazione plasmatica aumenta in modo circa esponenziale all’aumentare della dose (es. fenitoina).
.")
21
Legame con le proteine plasmatiche
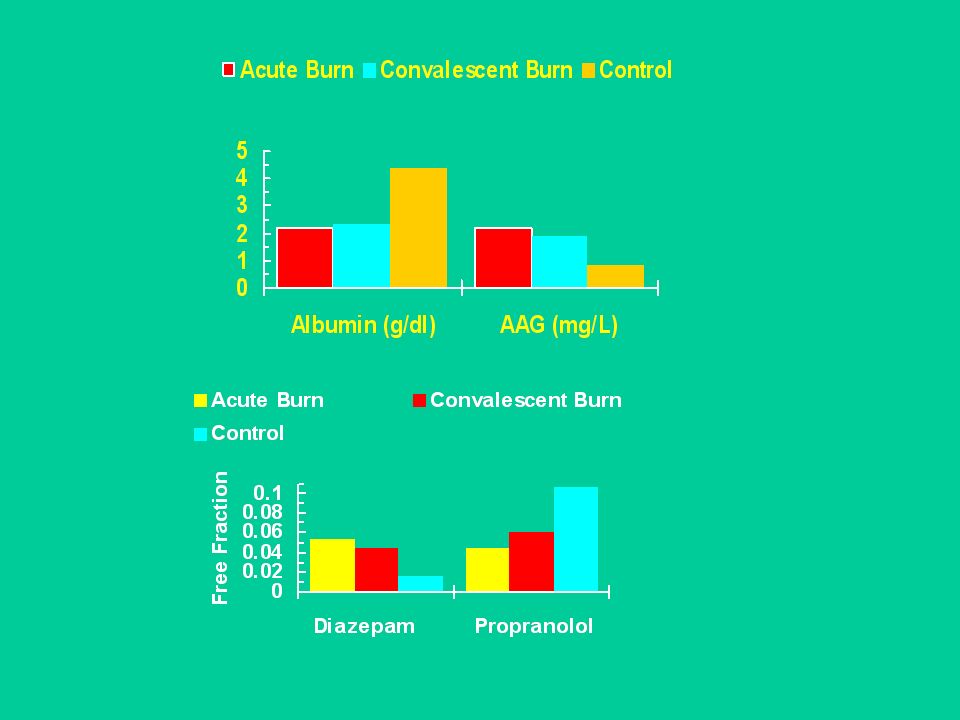
Nel sangue sono presenti molte proteine; la più abbondante è l’albumina. Queste proteine trasportano nel sangue sostanze normalmente presenti nell’organismo (acidi grassi, bilirubina ecc.) Molti farmaci si legano reversibilmente a siti di legame delle proteine plasmatiche, in particolare all’albumina. L’albumina lega prevalentemente farmaci acidi. Farmaci basici si legano alla ß-globulina e ad alla 1-glicoproteina acida.
 Molti farmaci si legano reversibilmente a siti di legame delle proteine plasmatiche, in particolare all’albumina. L’albumina lega prevalentemente farmaci acidi. Farmaci basici si legano alla ß-globulina e ad alla 1-glicoproteina acida.")
23
Il farmaco libero è in equilibrio con il farmaco legato:
F + P FP. Solo il farmaco libero diffonde nei tessuti ed esercita la sua attività*. Il legame alle proteine diminuisce quindi la concentrazione efficace di farmaco. Il legame con le proteine rallenta l’escrezione renale (a meno che questa avvenga per trasporto tubulare), perché solo la quota libera passa nel filtrato glomerulare. Le proteine plasmatiche funzionano come sito di deposito del farmaco concentrazioni attive di farmaco permangono anche dopo che è stata interrotta la somministrazione.
, perché solo la quota libera passa nel filtrato glomerulare. Le proteine plasmatiche funzionano come sito di deposito del farmaco concentrazioni attive di farmaco permangono anche dopo che è stata interrotta la somministrazione.")
24
* Se il farmaco si lega alle proteine tissutali, può essere rimosso dal sangue anche se legato alle proteine plasmatiche.

25
Concentrazione, quota legata, competizione
Consideriamo l’equilibrio: F + P FP Secondo l’equazione tipo Michaelis-Menten, la frazione di farmaco legato [B]/[F]+[B] dipende da: Concentrazione del farmaco libero, [F] Concentrazione della proteina plasmatica, [P] Numero di siti di legame per molecola di proteina, n costante di dissociazione, k (affinità di legame)
")
26
[B]/[F]+[B] = 1 1 + [F] + k n x [P]
Tuttavia, per la maggior parte dei farmaci, [P] >> [F], alle normali concentrazioni terapeutiche. La frazione di farmaco legato è quindi indipendente dalla concentrazione di farmaco ma dipende sostanzialmente da k/n x [P], cioè dall’affinità. All’aumentare della concentrazione di farmaco libero aumenta anche, proporzionalmente, la concentrazione di farmaco legato.
![[B]/[F]+[B] = [F] + k n x [P]](http://slideplayer.it/slide/938738/2/images/26/%5BB%5D%2F%5BF%5D%2B%5BB%5D+%3D+%5BF%5D+%2B+k+n+x+%5BP%5D.jpg "Tuttavia, per la maggior parte dei farmaci, [P] >> [F], alle normali concentrazioni terapeutiche. La frazione di farmaco legato è quindi indipendente dalla concentrazione di farmaco ma dipende sostanzialmente da k/n x [P], cioè dall’affinità. All’aumentare della concentrazione di farmaco libero aumenta anche, proporzionalmente, la concentrazione di farmaco legato.")
27
Per farmaci le cui concentrazioni terapeutiche sono vicine alla saturazione dei siti di legame, un aumento della dose (concentrazione totale del farmaco) provoca un aumento sproporzionato della concentrazione della quota libera (es. fenilbutazone). Dato che solo il farmaco libero è attivo, si potrebbe avere in questi casi la comparsa di tossicità. Tuttavia, l’aumento della concentrazione plasmatica del farmaco libero è seguito da un aumento della velocità di eliminazione (cinetica di I ordine)
")
28
In alcune condizioni patologiche (es
In alcune condizioni patologiche (es. epatopatie, stati inifammatori) la concentrazione delle proteine plasmatiche può variare. Di conseguenza, cambia anche la concentrazione di farmaco libero.
 la concentrazione delle proteine plasmatiche può variare. Di conseguenza, cambia anche la concentrazione di farmaco libero.")
30
Alcuni farmaci hanno concentrazioni terapeutiche tali da saturare i siti di legame con le proteine plasmatiche. Questi farmaci possono ‘spiazzare’ altri farmaci legati agli stessi siti.

31
Distribuzione ed accumulo
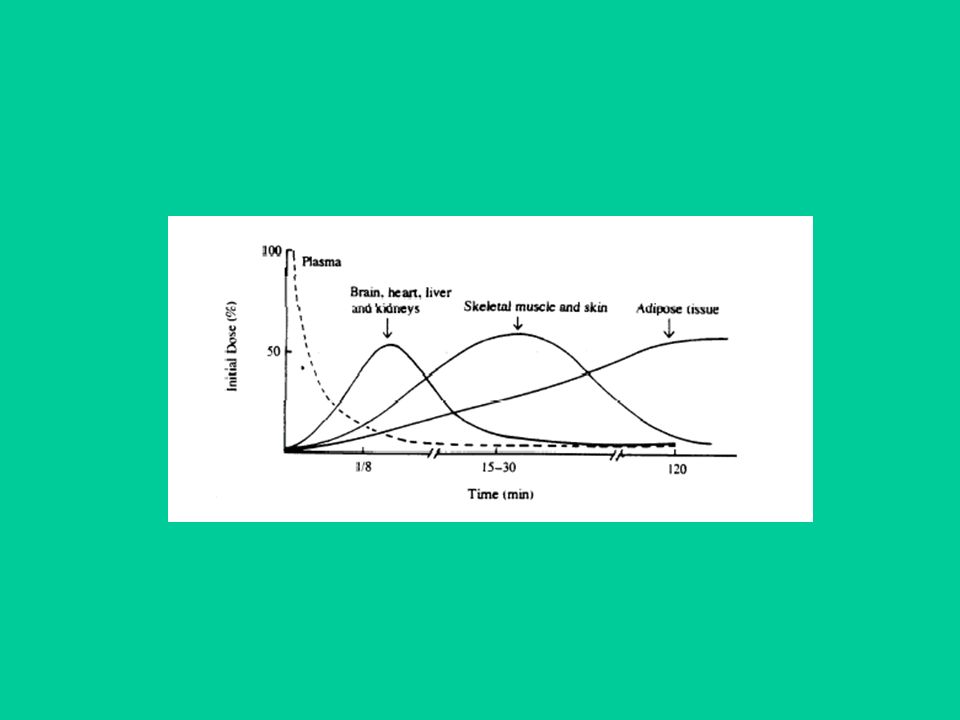
Una volta nel sangue, il farmaco (libero) va incontro a due processi: distribuzione ed eliminazione. Assorbimento, distribuzione ed eliminazione avvengono contemporaneamente. Il processo di distribuzione ai tessuti è, quasi sempre, più veloce del processo di eliminazione. La velocità di distribuzione iniziale ai vari tessuti, per un dato farmaco, dipende da: 1) permeabilità capillare nei tessuti; 2) flusso ematico nel tessuto.
 va incontro a due processi: distribuzione ed eliminazione. Assorbimento, distribuzione ed eliminazione avvengono contemporaneamente. Il processo di distribuzione ai tessuti è, quasi sempre, più veloce del processo di eliminazione. La velocità di distribuzione iniziale ai vari tessuti, per un dato farmaco, dipende da: 1) permeabilità capillare nei tessuti; 2) flusso ematico nel tessuto.")
32
Inizialmente si raggiungono concentrazioni elevate in organi altamente perfusi (es. fegato, reni, polmoni). La distribuzione finale dipende soprattutto dall’affinità della sostanza per i diversi tessuti. Diverse sostanze possono quindi ri-distribuirsi da tessuti ad alta perfusione a tessuti a bassa perfusione (es. tessuto adiposo, tessuto osseo) ma con più elevata affinità per il farmaco.
 ma con più elevata affinità per il farmaco.")
34
Ad esempio, 5 minuti dopo la somministrazione di TCDD (altamente lipofilo), il 15% della dose si ritrova nei polmoni e solo l’1% nel tessuto adiposo. Dopo 24 ore, il 20% della dose residua si ritrova nel tessuto adiposo, nei polmoni solo lo 0,3%. Il piombo si accumula (50% della dose) nel fegato 2 ore dopo la somministrazione. Dopo 1 mese, tuttavia, il 90% della dose residua si trova nel tessuto osseo.
 nel fegato 2 ore dopo la somministrazione. Dopo 1 mese, tuttavia, il 90% della dose residua si trova nel tessuto osseo.")
35
Alcune sostanze si accumulano in alcuni tessuti in conseguenza di alta liposolubilità, legame con proteine tissutali, trasporto attivo. Spesso il tessuto di deposito non è il tessuto bersaglio. In questo caso l’accumulo ha un effetto protettivo poiché diminuisce le concentrazioni nel plasma e nell’organo bersaglio. Tuttavia, il tossico viene reimmesso nel sangue man mano che la quota in circolazione viene eliminata permanenza di basse concentrazioni plasmatiche per lungo tempo.

36
Tessuti di deposito Proteine plasmatiche. Principalmente albumina Fegato e rene. Alcune proteine legano tossici (anioni organici, metalli pesanti) con alta affinità. La captazione avviene anche tramite sistemi di trasporto. Tessuto adiposo. Per sostanze lipofile. Tessuto osseo. Anioni e cationi (F, Pb, Sr) possono essere incorporati nella matrice ossea.
 con alta affinità. La captazione avviene anche tramite sistemi di trasporto. Tessuto adiposo. Per sostanze lipofile. Tessuto osseo. Anioni e cationi (F, Pb, Sr) possono essere incorporati nella matrice ossea.")
37
Le sostanze lipofile si accumulano nel tessuto adiposo
Le sostanze liposolubili, compresi i gas, si distribuiscono nei compartimenti lipidici dell’organismo. Il tessuto adiposo è poco irrorato e si equilibra quindi lentamente con il sangue. Può però agire da deposito per queste sostanze, rilasciandone quantità rilevanti nel sangue dopo la fine dell’esposizione.

38
Sostanze lipofile si distribuiscono rapidamente nel SNC, che ha un flusso ematico abbastanza elevato (350 ml/min). Il SNC, inoltre, è ricco di lipidi. Le sostanze lipofile si concentrano quindi nel SNC, oltre che nel tessuto adiposo.

39
Volume apparente di distribuzione (Vd)
Dato che il processo di distribuzione avviene in genere più velocemente di quello di eliminazione, si può estrapolare al concentrazione plasmatica (C0) alla fine della distribuzione (e prima che sia cominciata l’eliminazione). La quantità di farmaco presente nell’organismo in questo momento è data dalla dose somministrata (somministrazione e.v.) Dividendo la dose per la C0, si ottiene il volume che occuperebbe il farmaco se la concentrazione in tutti i fluidi dell’organismo fosse uguale a quella plasmatica. Per diversi farmaci, la conc. plasmatica è effettivamente uguale a quella a quella degli altri fluidi (liquidi extra- ed intracellulari). Per questi farmaci il volume di distribuzione è uguale al volume totale dei liquidi corporei (~ 0,6 l/kg; 42 l)
 alla fine della distribuzione (e prima che sia cominciata l’eliminazione). La quantità di farmaco presente nell’organismo in questo momento è data dalla dose somministrata (somministrazione e.v.) Dividendo la dose per la C0, si ottiene il volume che occuperebbe il farmaco se la concentrazione in tutti i fluidi dell’organismo fosse uguale a quella plasmatica. Per diversi farmaci, la conc. plasmatica è effettivamente uguale a quella a quella degli altri fluidi (liquidi extra- ed intracellulari). Per questi farmaci il volume di distribuzione è uguale al volume totale dei liquidi corporei (~ 0,6 l/kg; 42 l)")
40
Vd = D/C0 Farmaci che non attraversano le membrane cellulari rimangono confinati nell’acqua extracellulare. Per questi farmaci, la conc. plasmatica è maggiore rispetto ai farmaci che si distribuiscono in tutti i liquidi corporei; di conseguenza il loro Vd è minore (Vd 0,2 l/kg; 14 l). Ancora minore è il Vd di farmaci che rimangono confinati nel sangue (Vd 0,04 l/kg; 3 l) Per farmaci che si accumulano in uno o più tessuti, la conc. plasmatica è (molto) più bassa rispetto alla conc. di farmaci distribuiti in tutti i liquidi. Questi farmaci hanno valori di Vd maggiori del valore del volume totale dei liquidi corporei (es., clorochina 130 l/kg; l). Il volume di distribuzione è quindi, in generale, un volume apparente, che può o meno corrispondere al volume reale di un compartimento.
. Ancora minore è il Vd di farmaci che rimangono confinati nel sangue (Vd 0,04 l/kg; 3 l) Per farmaci che si accumulano in uno o più tessuti, la conc. plasmatica è (molto) più bassa rispetto alla conc. di farmaci distribuiti in tutti i liquidi. Questi farmaci hanno valori di Vd maggiori del valore del volume totale dei liquidi corporei (es., clorochina 130 l/kg; l). Il volume di distribuzione è quindi, in generale, un volume apparente, che può o meno corrispondere al volume reale di un compartimento.")
41
Importanza di Vd Vd può fornire indicazioni sulla distribuzione di una sostanza e sull’eventuale presenza di fenomeni di accumulo. Vd ha, inoltre, una grande importanza pratica in farmacologia. Infatti, se si vuole raggiungere una data concentrazione efficace di farmaco, la dose da somministrare è data da: D = Ceff x Vd.

42
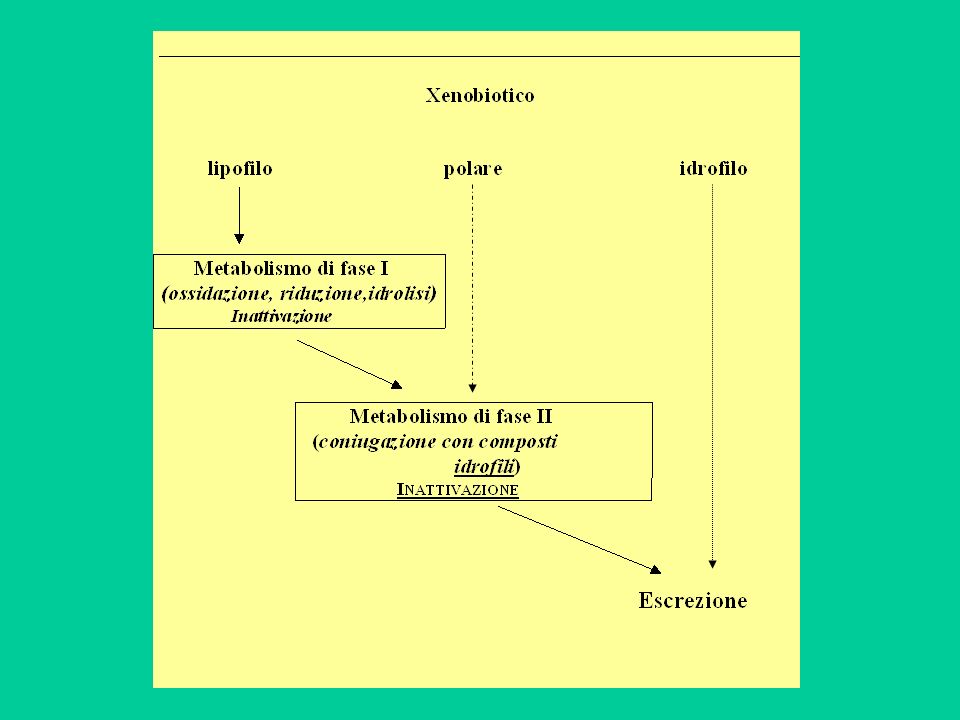
Metabolismo Il metabolismo di un farmaco (di uno xenobiotico) è l’insieme delle trasformazioni chimiche che la molecola subisce all’interno dell’organismo, principalmente ad opera di enzimi. Il metabolismo ha una ‘logica’: trasformare la molecola in modo che possa essere facilmente escreta per via renale o biliare. Dato che le molecole idrofile sono eliminate più rapidamente di quelle lipofile, il metabolismo consiste principalmente nella trasformazione di molecole lipofile in molecole idrofile.
 è l’insieme delle trasformazioni chimiche che la molecola subisce all’interno dell’organismo, principalmente ad opera di enzimi. Il metabolismo ha una ‘logica’: trasformare la molecola in modo che possa essere facilmente escreta per via renale o biliare. Dato che le molecole idrofile sono eliminate più rapidamente di quelle lipofile, il metabolismo consiste principalmente nella trasformazione di molecole lipofile in molecole idrofile.")
44
Le sostanze lipofile sono escrete più lentamente di quelle idrofile perché:
sono più facilmente riassorbite nel tubulo renale (o nell’intestino in caso di eliminazione biliare); si accumulano nel tessuto adiposo
; si accumulano nel tessuto adiposo.")
45
Durante il processo di formazione delle urine, si ha riassorbimento di acqua aumento della concentrazione dei soluti nell’urina gradiente di concentrazione urina-plasma riassorbimento. Le sostanze lipofile sono riassorbite più facilmente perché attraversano meglio lo strato cellulare dell’epitelio tubulare.

46
E’ stato calcolato che il tempo di dimezzamento (emivita) dell’esobarbitale, che è pari a 5-6 ore, sarebbe, in assenza di trasformazione metabolica, di 2-5 mesi.
 dell’esobarbitale, che è pari a 5-6 ore, sarebbe, in assenza di trasformazione metabolica, di 2-5 mesi.")
47
Variabilità della capacità metabolica
In natura, gli xenobiotici con cui vengono in contatto gli organismi animali sono di origine vegetale. La capacità di diverse specie animali di metabolizzare gli xenobiotici dipende dalla selezione operata dall’esposizione agli xenobiotici vegetali. Ad es., insetti che si cibano di molte piante diverse possono metabolizzare una varietà di xenobiotici maggiore degli insetti che si alimentano di poche o una sola specie vegetale. I pesci hanno minore capacità metabolizzante dei mammiferi, probabilmente perché possono più facilmente eliminare gli xenobiotici tramite le branchie.

48
L’evoluzione degli isozimi CYP450 riflette l’esposizione ad un numero sempre maggiore di xenobiotici potenzialmente tossici, sintetizzati dai vegetali a scopo difensivo.

49
Specie diverse di mammiferi possono avere differenze quali-quantitative della capacità metabolizzante importanti per l’estrapolazione all’uomo di dati animali. A livello individuale, la capacità metabolizzante è determinata, oltre che da fattori genetici, anche da fattori ambientali; la quantità di enzima presente può essere infatti aumentata da alcune sostanze (induzione enzimatica).
.")
50
Caratteristiche degli enzimi metabolizzanti
Gli enzimi metabolizzanti sono in genere poco specifici, metabolizzano cioè molti substrati diversi (ma con un dominio strutturale comune). Alcuni enzimi sono deputati solo al metabolismo degli xenobiotici, altri sono coinvolti nella sintesi o degradazione di composti endogeni. L’organo più ricco di enzimi metabolizzanti è il fegato. Altri organi o tessuti con significativa capacità metabolica sono i polmoni, i reni, il sangue. Alcuni tessuti hanno un’elevata concentrazione di enzimi metabolizzanti (mucosa nasale, cristallino), ma il loro contributo al metabolismo sistemico è pressoché nullo dato il loro piccolo volume.
. Alcuni enzimi sono deputati solo al metabolismo degli xenobiotici, altri sono coinvolti nella sintesi o degradazione di composti endogeni. L’organo più ricco di enzimi metabolizzanti è il fegato. Altri organi o tessuti con significativa capacità metabolica sono i polmoni, i reni, il sangue. Alcuni tessuti hanno un’elevata concentrazione di enzimi metabolizzanti (mucosa nasale, cristallino), ma il loro contributo al metabolismo sistemico è pressoché nullo dato il loro piccolo volume.")
51
La localizzazione degli enzimi è importante nel determinare l’organo colpito nel caso di formazione di metaboliti tossici. Molti metaboliti tossici sono infatti altamente reattivi e colpiscono quindi solo le cellule dell’organo in cui si formano. Ad es., molti composti sono epatotossici perché i metaboliti reattivi si formano nel fegato. Inoltre, la tossicità è spesso limitata, all’interno dell’organo, alle cellule che possiedono l’enzima che porta alla formazione del metabolita tossico. Ad es., il paracetamolo e CCl4 causano necrosi centrolobulare perché le cellule della regione centrolobulare sono ricche di citocromo P450.

52
Localizzazione intracellulare degli enzimi
Un determinato enzima ha in genere un’unica localizzazione intracellulare. Nel fegato gli enzimi sono localizzati principalmente nel reticolo endoplasmatico (enzimi microsomiali) e nel citosol. Un numero minore di enzimi è localizzato nei mitocondri, nei lisosomi, nel nucleo.
 e nel citosol. Un numero minore di enzimi è localizzato nei mitocondri, nei lisosomi, nel nucleo.")
53
Reazioni di fase I Sono le reazioni di idrolisi, riduzione, ossidazione. Portano in genere all’introduzione o smascheramento di un gruppo nucleofilo (-OH, -NH2, -SH, -COOH). Ciò causa solo un modesto aumento dell’idrofilia. Tuttavia, il gruppo funzionale nucleofilo fornisce un punto di attacco per le reazioni di fase II. Le reazioni di fase I determinano in genere perdita dell’attività farmaco-tossicologica (modificazione della struttura chimica e della capacità di interagire con il recettore). In alcuni casi, tuttavia, i prodotti delle reazioni di fase I sono biologicamente attivi. I pro-farmaci sono attivati dalle reazioni di fase I. Nelle reazioni di fase I, soprattutto le reazioni di ossidazione, si possono formare metaboliti tossici.
. Ciò causa solo un modesto aumento dell’idrofilia. Tuttavia, il gruppo funzionale nucleofilo fornisce un punto di attacco per le reazioni di fase II. Le reazioni di fase I determinano in genere perdita dell’attività farmaco-tossicologica (modificazione della struttura chimica e della capacità di interagire con il recettore). In alcuni casi, tuttavia, i prodotti delle reazioni di fase I sono biologicamente attivi. I pro-farmaci sono attivati dalle reazioni di fase I. Nelle reazioni di fase I, soprattutto le reazioni di ossidazione, si possono formare metaboliti tossici.")
54
Enzimi di fase I- Reazioni di Idrolisi
Carbossilesterasi (esterasi). Idrolisi di esteri (veloce), amidi (lenta) esteri tiolici, esteri fosforici, anidridi acide. Es.: RCOOR1 + H2O RCOOH + R1OH Le esterasi sono enzimi poco specifici. Oltre agli xenobiotici, metabolizzano anche composti endogeni. Sono ampiamente distribuite nell’organismo; i livelli più alti si trovano nel fegato, plasma ed eritrociti, rene (tubulo prossimale), polmone.
. Idrolisi di esteri (veloce), amidi (lenta) esteri tiolici, esteri fosforici, anidridi acide. Es.: RCOOR1 + H2O RCOOH + R1OH. Le esterasi sono enzimi poco specifici. Oltre agli xenobiotici, metabolizzano anche composti endogeni. Sono ampiamente distribuite nell’organismo; i livelli più alti si trovano nel fegato, plasma ed eritrociti, rene (tubulo prossimale), polmone.")
55
Peptidasi e proteasi: Endopeptidasi: idrolizzano un legame peptidico interno. Esopeptidasi: idrolizzano un legame peptidico terminale (carbossipeptidasi, aminopeptidasi); alcune ‘staccano’ 2 o 3 aminoacidi terminali (di- o tri-peptidil-peptidasi). Largamente presenti nel tratto GI. Negli altri tessuti, possono essere ancorate alla membrana cellulare o essere secrete all’esterno della cellula; sono presenti anche nel sangue.
; alcune ‘staccano’ 2 o 3 aminoacidi terminali (di- o tri-peptidil-peptidasi). Largamente presenti nel tratto GI. Negli altri tessuti, possono essere ancorate alla membrana cellulare o essere secrete all’esterno della cellula; sono presenti anche nel sangue.")
56
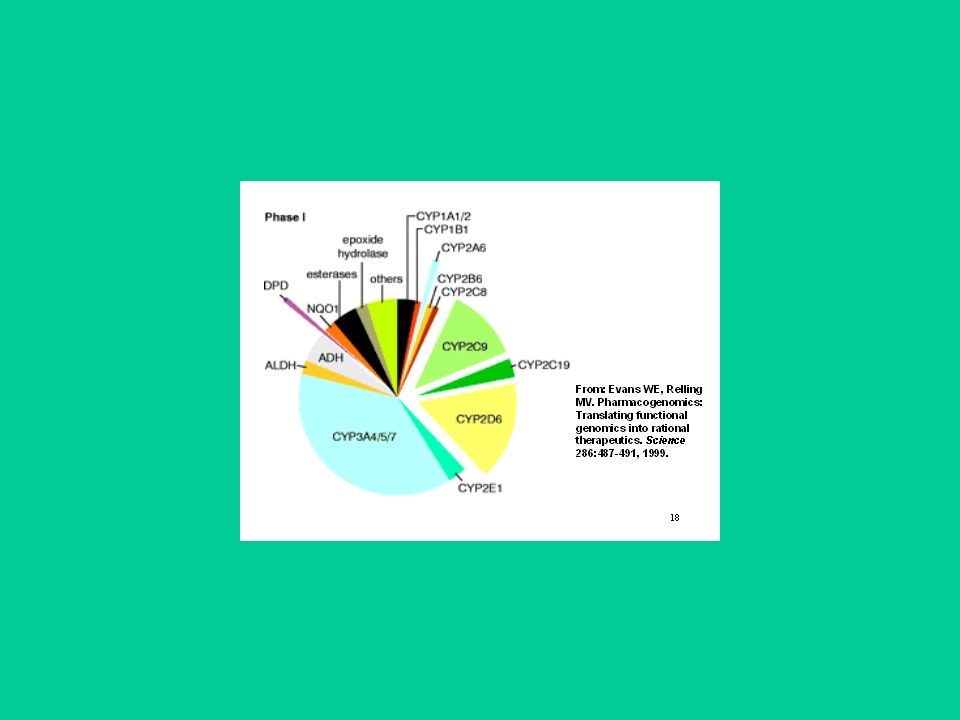
Enzimi di fase I - Ossidazioni
Citocromo P450 (CYP 450). E’ l’enzima più importante di fase I perché metabolizza un gran numero di xenobiotici principale responsabile dell’inattivazione di molti farmaci e tossici diretti. Coinvolto anche nella biosintesi o degradazione di molti composti endogeni (ormoni steroidei, vitamine liposolubili, acidi grassi ecc.) Presente in tutti i tessuti. I livelli più alti sono nel fegato, a livello del reticolo endoplasmatico (frazione microsomiale)
. E’ l’enzima più importante di fase I perché metabolizza un gran numero di xenobiotici principale responsabile dell’inattivazione di molti farmaci e tossici diretti. Coinvolto anche nella biosintesi o degradazione di molti composti endogeni (ormoni steroidei, vitamine liposolubili, acidi grassi ecc.) Presente in tutti i tessuti. I livelli più alti sono nel fegato, a livello del reticolo endoplasmatico (frazione microsomiale)")
58
Reazione generale: RH (substrato) + O2 + NADPH + H+ ROH H2O + NADP+ E’ una reazione di monoossigenazione in cui un atomo di ossigeno è incorporato nel substrato; l’altro atomo di O è ridotto ad H2O (CYP450 è quindi un’ossidasi a funzione mista), con l’apporto degli equivalenti riducenti (elettroni) provenienti da NADPH. Il prodotto ROH può subire riarrangiamenti molti prodotti finali, diversi da alcol e fenoli.
, con l’apporto degli equivalenti riducenti (elettroni) provenienti da NADPH. Il prodotto ROH può subire riarrangiamenti molti prodotti finali, diversi da alcol e fenoli.")
59
amino ossidasi Tipi di reazione catalizzate dal CYP450:
Idrossilazione alifatica e aromatica De-alchilazione di un eteroatomo Ossigenazione di un eteroatomo Trasferimento di un gruppo ossidativo. Riduzione

60
Epossidazione

61
Epossido idrolasi. Catalizzano l’addizione di H2O agli epossidi formati (dal citocromo P450) nelle reazioni di ossidazione di alcheni e composti arilici. Dato che questi epossidi sono quasi tutti altamente reattivi e tossici (cancerogeni), le epossido idrolasi sono enzimi detossificanti molto importante. Sono ampiamente distribuiti nell’organismo; all’interno di alcuni tessuti, la loro distribuzione è parallela a quelle del citocromo P450 rapida detossificazione degli epossidi. Alcuni epossidi non possono essere metabolizati dall’epossido idrolasi per ragioni steriche (bay region) e sono quindi particolarmente tossici (es., benzopirene-9,10-epossido).
, le epossido idrolasi sono enzimi detossificanti molto importante. Sono ampiamente distribuiti nell’organismo; all’interno di alcuni tessuti, la loro distribuzione è parallela a quelle del citocromo P450 rapida detossificazione degli epossidi. Alcuni epossidi non possono essere metabolizati dall’epossido idrolasi per ragioni steriche (bay region) e sono quindi particolarmente tossici (es., benzopirene-9,10-epossido).")
62
L’epossido idrolasi è un enzima inducibile
L’epossido idrolasi è un enzima inducibile. La sua induzione è sempre associata a quella del citocromo P450. Può essere inibito da alcuni epossidi e da alcuni farmaci aumento della tossicità di composti metabolizzati ad epossidi.

Presentazioni simili


















 che regola la por tata di aria che.>")



