Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Malattie della mielina

2
Sclerosi Multipla Patologia demielinizzante infiammatoria multifocale del SNC a verosimile patogenesi autoimmune

3
1. Malattie demielinizzanti (SM)
Distruzione della mielina normalmente costituita 2. Malattie dismielinizzanti Errore di sintesi o di metabolismo

4
La mielina 75% lipidi: 20-25% proteine
PLP (proteina proteolipidica) 50% MBP (proteina basica della mielina) 30-35% MAG (Glicoproteina associata alla mielina) 0.5% MOG (glicoproteina mielinica oligodendrocitaria.) 0.05% a B-cristallina CNPasi (2’3’nucleotide ciclica-fosfodiesterasi)
 50% MBP (proteina basica della mielina) 30-35% MAG (Glicoproteina associata alla mielina) 0.5% MOG (glicoproteina mielinica oligodendrocitaria.) 0.05% a B-cristallina. CNPasi (2’3’nucleotide ciclica-fosfodiesterasi)")
5
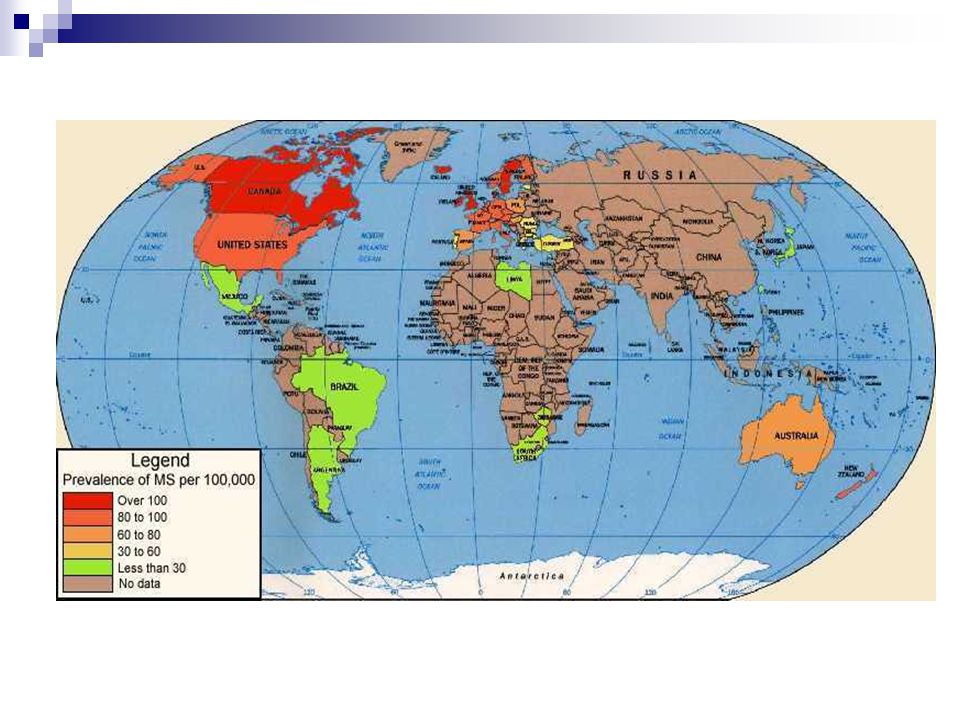
Epidemiologia Prevalenza Nord America >200/100,000
Nord Europa 100/100,000 Regioni tropicali 5/100,000 Italia 50/100,000 (incidenza 2-4 casi 100,000 abitanti per anno) F/M = 2-3/1 Picco massimo di incidenza 30 anni
 F/M = 2-3/1. Picco massimo di incidenza 30 anni.")
6
>30 casi 5-25 casi < 5 casi

8
Europa centro-settentrionale (>46° latitudine)
Prevalenza Incidenza Casi/100, Casi/100,000 Norvegia: Islanda Finlandia Svezzia UK Olanda Germania Polonia Cecoslovaccha / Ungheria Svizzera /

9
Europa Meridionale (36°-46° latitudine)
Prevalenza Incidenza Casi/100, Casi/100,000 Bulgaria / Croazia Francia / Grecia Romania / Slovenia Spagna Italia continentale Sardegna Sicilia

10
Da G. Rosati: The prevalence of multiple sclerosis in the world: an update
Neurol Sci (2001) 22:
 22:")
11
SM in Sicilia Enna (Dean G. 1975) 53.2 (1975) /
Prevalenza (PD) Incidenza Enna (Dean G. 1975) (1975) / Enna (Grimaldi L.2001) (1995) (86-95) Caltanissetta (Savettieri G. 1981) (1981) / Agrigento (Dean G. 1975) (1975) / Monreale (Savettieri G. 1980) (1980) / Monreale (Savettieri G .1991) (1991) (81-91) Catania (Nicoletti A. 2001) (1995) (75-95) Catania (Nicoletti A. ****) (2000) (90-00) Bagheria (Savettieri G .2000) / (85-94)
 Incidenza. Enna (Dean G. 1975) 53.2 (1975) / Enna (Grimaldi L.2001) 120 (1995) 5.7 (86-95) Caltanissetta (Savettieri G. 1981) 51.0 (1981) / Agrigento (Dean G. 1975) 32.0 (1975) / Monreale (Savettieri G. 1980) 43.3 (1980) / Monreale (Savettieri G .1991) 72.4 (1991) 3.3 (81-91) Catania (Nicoletti A. 2001) 58.5 (1995) 2.3 (75-95) Catania (Nicoletti A. ****) 90.0 (2000) 5.5 (90-00) Bagheria (Savettieri G .2000) / 4.4 (85-94)")
12
Eziopatogenesi Fattori genetici HLA-A3, B7, -DR2, -DQW1
Fattori ambientali (migrazione) Ipotesi virale (morbillo, parotite, Herpes, retrovirus, JC etc)
 Ipotesi virale (morbillo, parotite, Herpes, retrovirus, JC etc)")
13
Anatomia patologica Placca acuta attiva = edema e flogosi
Placca cronica attiva = demielinizzazione Placca cronica silente = gliosi astrocitaria E caratteristica la presenza di un infiltrato infiammatorio con cellule di tipi istiocitico-macrofagico, linfociti T e plasmacellule in sede perivascolare ed alla periferia della placca attiva, acuta o cronica.

14
Placche di demielinizzazione
Sede = corpo calloso e sb peiventricolare e cervelletto Sede perivasale Da meno di un millimetro a qualche centimetro

15
Eziopatogenesi Attivazione dei linfociti T (CD8+) al livello periferico Migrazione attraverso la BEE Formazione di infiltrato infiammatorio Danno alla mileina

16
Demyelination and Axon Loss Danger Signal or Trigger
MS Pathogenesis CNS Autoreactive T Cells T Periphery Demyelination and Axon Loss BBB Transmigration Danger Signal or Trigger Activation, Differentiation, Clonal Expansion Local Reactivation APC Adhesion/Attraction Release of Cytokines; Recruitment of M Antibodies B M NO IFN- TNF- MS Pathogenesis Autoreactive T cells in the periphery are activated by a danger signal or trigger. They migrate to, adhere at, and penetrate through the BBB, steps mediated by adhesion molecules, proteases, and chemokines.1 Inside the CNS, the T cells are reactivated by APCs, predominantly microglial cells. The reactivated T cells secrete pro-inflammatory cytokines, such as IFN- or IL-2, which induce CNS inflammation by subsequent activation of macrophages such as other T cells and B cells. Macrophages and T cells attack the myelin sheath of oligodendrocytes by cytotoxic mediators, mainly TNF-, O2 radicals, and NO. B cells differentiate into plasma cells. These secrete demyelinating antibodies that can guide and activate macrophages (M) and ignite the complement cascade, which causes assembly of the membrane attack complex and pore formation in myelin membranes. Demyelination can occur by four different pathologic processes, including T-cell and macrophage attack, antibody attack, oligodendrocyte apoptosis, and primary oligodendrocyte degeneration.1 1. Neuhaus O, Archelos JJ, Hartung HP. Immunomodulation in multiple sclerosis: from immunosuppression to neuroprotection. Trends Pharmacol Sci ;24: Adapted with kind permission from Prof. R. Hohlfeld.
 and ignite the complement cascade, which causes assembly of the membrane attack complex and pore formation in myelin membranes. Demyelination can occur by four different pathologic processes, including T-cell and macrophage attack, antibody attack, oligodendrocyte apoptosis, and primary oligodendrocyte degeneration Neuhaus O, Archelos JJ, Hartung HP. Immunomodulation in multiple sclerosis: from immunosuppression to neuroprotection. Trends Pharmacol Sci. 2003;24: Adapted with kind permission from Prof. R. Hohlfeld.")
17
Patogenesi della SM Attivazione cellule T periferiche specifiche per PBM Linfociti T attivati aumentano l‘espressione delle molecole di adesione sulle cellule endoteliali della BEE (integrine, V-CAM), che consentono al linfocita di aderire alle cellule endoteliali. Processo mediato da citochine pro-infiammatorie TNFa INFg etc.) Legame delle integrine con le controparti presenti sui linfo-monociti (I-CAM) Liberazione di enzimi litici (metallo-proteasi, gelatinasi) Scissione delle giunzioni endoteliali Alterazione della BEE Diapedesi delle cellule immunitarie Cellule T attivate rilasciano citochine pro-infiammatorie (INFg, TNFa etc.) Macrofagi perivascolari e microglia agiscono come cellule presentanti l’antigene ai linfociti T (componenti mieliniche legate al complesso maggiore di istocompatibilità II) portando alla proliferazione dei Linfociti T. T citotossici, macrofagi, microglia producono citochine pro-infiammatorie che attaccano gli oligodendrociti. (Demielinizzazione) Cellule T stimolano le cellule B a produrre anticorpi contro i componenti della mielina Interazione antigene anticorpo, rilascio di complemento, stimolazione dei fagociti e lisi degli oligodendrociti Ulteriore alterazione della BEE
, che consentono al linfocita di aderire alle cellule endoteliali. Processo mediato da citochine pro-infiammatorie TNFa INFg etc.) Legame delle integrine con le controparti presenti sui linfo-monociti (I-CAM) Liberazione di enzimi litici (metallo-proteasi, gelatinasi) Scissione delle giunzioni endoteliali. Alterazione della BEE. Diapedesi delle cellule immunitarie. Cellule T attivate rilasciano citochine pro-infiammatorie (INFg, TNFa etc.) Macrofagi perivascolari e microglia agiscono come cellule presentanti l’antigene ai linfociti T (componenti mieliniche legate al complesso maggiore di istocompatibilità II) portando alla proliferazione dei Linfociti T. T citotossici, macrofagi, microglia producono citochine pro-infiammatorie che attaccano gli oligodendrociti. (Demielinizzazione) Cellule T stimolano le cellule B a produrre anticorpi contro i componenti della mielina. Interazione antigene anticorpo, rilascio di complemento, stimolazione dei fagociti e lisi degli oligodendrociti. Ulteriore alterazione della BEE.")
18
Mielina ed autoimmunità
75% lipidi: PLP (proteina proteolipidica) 50% Nel caso della Sm la risposta immunitaria nei confronti della PLP non sembra essere l’evento primario nella patogenesi della malattia MBP (proteina basica della mielina) 30-35% La risposta T linfocitaria alla MBP comporta una produzione citochinica di tipo Th1 (INF gamma, TNF alfa IL2) MAG (Glicoproteina associata alla mielina) 0.5% Non è implicata nella SM MOG (glicoproteina mielinica oligodendrocitaria.) 0.05% Da molti ritenuta il principale target primario dell’autoimmunità a B-cristallina CNPasi (2’3’nucleotide ciclica-fosfodiesterasi)
 50% Nel caso della Sm la risposta immunitaria nei confronti della PLP non sembra essere l’evento primario nella patogenesi della malattia. MBP (proteina basica della mielina) 30-35% La risposta T linfocitaria alla MBP comporta una produzione citochinica di tipo Th1 (INF gamma, TNF alfa IL2) MAG (Glicoproteina associata alla mielina) 0.5% Non è implicata nella SM. MOG (glicoproteina mielinica oligodendrocitaria.) 0.05% Da molti ritenuta il principale target primario dell’autoimmunità. a B-cristallina. CNPasi (2’3’nucleotide ciclica-fosfodiesterasi)")
19
Citochine e SM I linfociti T CD4+ e CD8+ T helper 1 (Th1)
secernono:IL-2 INFg TNF Stimolano la produzione di altre citochine Th1 Bloccano le Th2 Favoriscono l’espansione clonale di linfociti T Citotossici Mediano le reazione di ipersensibilità ritardata. T helper 2 (Th2) Producono: IL-4 IL-5 IL-10 e IL-13 Inducono la sintesi di IgE Mediano le reazioni allergiche
 Producono: IL-4 IL-5 IL-10 e IL-13. Inducono la sintesi di IgE. Mediano le reazioni allergiche.")
20
L La doppia natura dell’infiammazione nella SM
Pro-infiammatoria e fattori neurotossici Anti-infiammatoria e fattori neuroprotrttivi Th1 cytokines TNF- IL-1 Nitric oxide Reactive oxygen species Glutamate Antibodies and complement Cell-mediated neurotoxicity Th2 cytokines TGF- IL-1 Neurotrophic factors – BDNF – NGF – NT-3 – CNTF – GDNF The Dual Nature of Inflammation in MS Well-established treatments such as steroids (during relapses) and glatiramer acetate or interferon- all have an anti-inflammatory action. All of them affect relapses; glatiramer acetate and some interferon- treatments also slow progression of disability. However, it is worth considering that some components of the inflammatory response are probably beneficial. Recently, an increasing body of evidence has appeared that supports the concept of neuroprotective autoimmunity in MS—a dual role for the immune system in which inflammation is beneficial as well as harmful. DANNO TESSUTALE PROTEZIONE TESSUTALE
 and glatiramer acetate or interferon- all have an anti-inflammatory action. All of them affect relapses; glatiramer acetate and some interferon- treatments also slow progression of disability. However, it is worth considering that some components of the inflammatory response are probably beneficial. Recently, an increasing body of evidence has appeared that supports the concept of neuroprotective autoimmunity in MS—a dual role for the immune system in which inflammation is beneficial as well as harmful. DANNO TESSUTALE. PROTEZIONE TESSUTALE.")
21
MS: A Disease of Severe Myelin, Axonal, and Neuronal Losses
Normal White Matter Plaque Myelin Axons Astrocytes Neurons Lymphocytes Macrophages MS: A Disease of Severe Myelin, Axonal, and Neuronal Losses Neuronal damage is now recognized as a major cause of permanent disability in people with MS. Neuronal degeneration starts early in the disease. The role of axonal demyelination has been recognized for many years as a major and prominent hallmark of MS. In recent years, however, studies have established that early axonal degeneration is also a prominent feature of the disease, and that it correlates with the persistent accumulation of permanent neurologic deficit over time. The incidence of axonal damage and degeneration is known to be highest in actively demyelinating lesions in the presence of inflammation and cytotoxic T cells, but it continues at a “slow burn” (~1/100th the rate in active lesions) seemingly for months or years after the active period has subsided. Curiously, the magnitude of the slow burn appears to be related to the magnitude of the preceding inflammation, as if inflammation triggers a sequence of events that leads slowly to degeneration. Evidence from animal models implicates activated T cells in initiating the pathology of MS. Subsequent injury to the CNS is mediated by T cells, B cells, and macrophages/microglia. Inflammatory components destroy myelin and oligodendrocytes. Inflammation is associated with axonal damage Adapted with kind permission from Dr. W. Brück.
 seemingly for months or years after the active period has subsided. Curiously, the magnitude of the slow burn appears to be related to the magnitude of the preceding inflammation, as if inflammation triggers a sequence of events that leads slowly to degeneration. Evidence from animal models implicates activated T cells in initiating the pathology of MS. Subsequent injury to the CNS is mediated by T cells, B cells, and macrophages/microglia. Inflammatory components destroy myelin and oligodendrocytes. Inflammation is associated with axonal damage. Adapted with kind permission from Dr. W. Brück.")
22
Axonal Damage in MS Terminal axonal ovoids
Nonphosphorylated neurofilaments Axonal Damage in MS These confocal micrographs from postmortem tissue of MS patients were obtained in a very elegant study by Trapp and colleagues and show axonal damage and demyelination in active MS lesions.1 The sections were taken from MS lesions and were stained with fluorescent antibody to myelin (red) and nonphosphorylated neurofilaments (green), which are usually abundant only in neuronal cell bodies and dendrites. The presence of nonphosphorylated neurofilaments in the axon indicates demyelination. Panel A is from the center of an active lesion. The arrows point to terminal axonal ovoids with single axonal connections, and the arrowhead points to an axonal ovoid with two axonal connections. The scale bar equals 64 microns. Panel B shows three large axons that stained positively for nonphosphorylated neurofilaments and are undergoing active demyelination (arrowheads). The arrow points to a large terminal ovoid at the end of an axon. The scale bar equals 45 microns. 1. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338: 64 m 45 m Adapted with permission from Trapp BD et al. N Engl J Med. 1998;338:
 and nonphosphorylated neurofilaments (green), which are usually abundant only in neuronal cell bodies and dendrites. The presence of nonphosphorylated neurofilaments in the axon indicates demyelination. Panel A is from the center of an active lesion. The arrows point to terminal axonal ovoids with single axonal connections, and the arrowhead points to an axonal ovoid with two axonal connections. The scale bar equals 64 microns. Panel B shows three large axons that stained positively for nonphosphorylated neurofilaments and are undergoing active demyelination (arrowheads). The arrow points to a large terminal ovoid at the end of an axon. The scale bar equals 45 microns. 1. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338: m. 45 m. Adapted with permission from Trapp BD et al. N Engl J Med. 1998;338:")
23
Eziopatogenesi Fattori genetici HLA-A3, B7, -DR2, -DQW1
Fattori ambientali (migrazione) Ipotesi virale (morbillo, parotite, Herpes, retrovirus, JC etc)
 Ipotesi virale (morbillo, parotite, Herpes, retrovirus, JC etc)")
24
Infanzia Adolescenza Età adulta SM Conclamata
Suscettibilità Genetica Geni Multipli Età critica A B C Infanzia Adolescenza Età adulta Fattori A Fattori B Fattori C Trigger Fattori dell’ambiente fisico, biologico, sociale SM Conclamata Fattori in grado di modulare il decorso

25
INTERAZIONE MULTIFATTORIALE TRA INFLUENZE AMBIENTALI E GENETICO-RAZZIALI NELLA EZIOLOGIA DELLA SCLEROSI MULTIPLA: PRINCIPALI FATTORI DI RISCHIO Fattori intrinseci all’individuo Fattori dell’ambiente biologico Razza Agenti infettivi e virus in particolare Predisposizione genetica Dieta e nutrizione Età Traumi Sesso Interventi chirurgici Familiarità Contatti con animali Genitura Vaccinazioni-immunizzazioni Gravidanze Fattori dell’ambiente fisico Fattori dell’ambiente sociale Latitudine Classe sociale e scolarità Altitudine Condizioni igienico-sanitarie Clima Occupazione Agenti ambientali Esposizione ad agenti tossici Composizione delle acque e del suolo nell’ambiente di lavoro

26
Ipotesi infettiva: ruolo del EBV
US Cohort (US Army) Prospective nested case-control study (83 casi e 166 controlli) RR19.7 (95%CI ) (VCA) RR 33.9 (95% ) (EBNA) Nessuna associazione per CMV Nessuna associazione per Chlamydia pneumoniae* Levin LI et al., JAMA 2003 *Munger KL Neurology 2004
 Prospective nested case-control study. (83 casi e 166 controlli) RR19.7 (95%CI ) (VCA) RR 33.9 (95% ) (EBNA) Nessuna associazione per CMV. Nessuna associazione per Chlamydia pneumoniae* Levin LI et al., JAMA *Munger KL Neurology")
27
Ipotesi infettiva: ruolo del EBV
Northern Sweeden Health and Disease (NSHDS) Prospective nested case-control study (73* prospective cases; 161 retrospective cases) *OR 4.2 (95%CI ) (EBV-EBNA) *OR 2.3 (95%CI 1.0–5.1) (HH6) *OR 11 (95%CI ) (EBV-EBNA) (< 5 anni) *OR 12 (95%CI 1.8–76) (HH6) (< 5 anni) Nessuna associazione per HSV, VZV Morbillo, *Sundstrom Neurology 2004
 Prospective nested case-control study. (73* prospective cases; 161 retrospective cases) *OR 4.2 (95%CI ) (EBV-EBNA) *OR 2.3 (95%CI 1.0–5.1) (HH6) *OR 11 (95%CI ) (EBV-EBNA) (< 5 anni) *OR 12 (95%CI 1.8–76) (HH6) (< 5 anni) Nessuna associazione per HSV, VZV Morbillo, *Sundstrom Neurology")
28
Quadro clinico Interessamento dei nervi cranici Disturbi motori
Disturbi sensitivi Disturbi della coordinazione e dei movimenti Disturbi sfinterici e sessuali Altri

29
Sintomi d’esordio Ipostenia 30% Neurite Ottica 22%
Parestesie disestesie 18% Sintomi cerebellari % Diplopia vertigini, disturbi della minzione 10% Manifestazioni parossistiche o psichiche 5%

30
Esordio della malattia
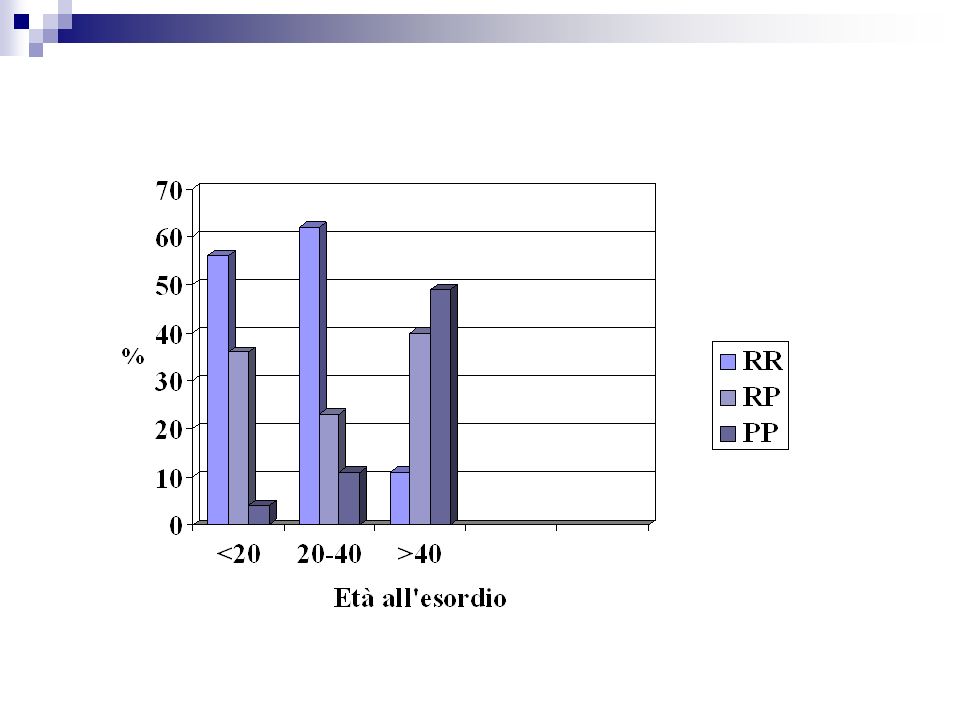
Disturbi motori Più frequente negli esordi tardivi (46% >40 anni) Disturbi sensitivi Meno frequente negli esordi precoci Disturbi cerebellari 15% Neurite ottica 20-25% <20 anni 16-18% anni Si riduce dopo i 40 anni Diplopia più frequente negli esordi precoci
 Disturbi sensitivi. Meno frequente negli esordi precoci. Disturbi cerebellari 15% Neurite ottica % <20 anni % anni. Si riduce dopo i 40 anni. Diplopia. più frequente negli esordi precoci.")
31
Altri sintomi d’esordio (<10%)
Nevralgia del trigemino Disturbi parossistici di altra natura Lesioni dei nervi cranici oltre n.ottici ed oculomotori Manifestazioni epilettiche Disturbi urogenitali Disturbi delle funzioni cognitive e della sfera affettiva

32
Modalità d’esordio Monosintomatico (2/3 casi);
più frequente negli esordi tardivi (paraparesi spastica progressiva) Acuto-Subacuto (70% dei casi ad esordio giovanile) Progressivo (frequente negli esordi tardivi)
 Acuto-Subacuto. (70% dei casi ad esordio giovanile) Progressivo. (frequente negli esordi tardivi)")
33
Evoluzione Fatica Disturbi urogenitali
Disturbi intestinali e della funzione sfinterica anale Dolori Epilessia Disturbi parossistici Disartria e disfagia

34
Che cos’è una esacerbazione o ricaduta
Comparsa di nuovi sintomi o ricomparsa di sintomi pregressi Aggravamento di sintomi preesistenti Durata superiore alle 24 ore Le ricadute devono essere separate da almeno 1 mese Esclusione di pseudoattacchi Frequenza delle ricadute 0.6 annue (0.9 RR e 0.6 RP)
")
35
Decorso Ricadute e remissioni (RR) 30%
Progressiva e a ricadute (PR) 10% Secondariamente progressiva (SP) 25% Primariamente –progressiva (PP) <20% Varianti benigne % (EDSS 3.5 dopo anni) Varianti maligne 5%
 10% Secondariamente progressiva (SP) 25% Primariamente –progressiva (PP) <20% Varianti benigne 15% (EDSS 3.5 dopo anni) Varianti maligne 5%")
37
Passaggio a forme secondariamente progressiva
Età media 38 anni 1-5 anni dall’esordio: % SP 6-10 anni dall’esordio: 41.3% 11-15 anni dall’esordio: 57.6%

38
Expanded Disability Status Scale (EDSS)
Sistemi Funzionali Punteggi FP (piramidale) FC (cerebellare) FT (Trocoencefalico) 0-5 FS (Sensitivo) FSf (Sfinterico) FV (Visivo) FM (Mentale) Altro* Spasticità EDSS
 0-6. FC (cerebellare) 0-5. FT (Trocoencefalico) 0-5. FS (Sensitivo) 0-6. FSf (Sfinterico) 0-6. FV (Visivo) 0-6. FM (Mentale) 0-5. Altro* 0-1. Spasticità 0-3. EDSS")
39
Disabilità E’ variabile ma in relazione alla durata di malattia.
EDSS = 3 (segni di disfunzione neurologica senza effetti sull’autonomia) 1/3 casi tra anni 1/3 casi entro i 2 anni 1/3 casi oltre i 7 anni (Mediamente entro i 7 anni ) EDSS = 6 (severa limitazione dell’autonomia) 9-15 anni.
 1/3 casi tra 2-7 anni. 1/3 casi entro i 2 anni. 1/3 casi oltre i 7 anni. (Mediamente entro i 7 anni ) EDSS = 6 (severa limitazione dell’autonomia) 9-15 anni.")
40
Inflammation and Axonal Loss in MS
Repairing-Remitting Secondary Progression Clinical Disability Clinical Threshold Brain Volume Inflammation and Axonal Loss in MS The above figure illustrates the course of MS.1-5 During the relapsing-remitting phase, the disease is characterized by frequent inflammation, demyelination, axonal transection, and remyelination. Relapses are more frequent and complete recovery from disability generally occurs.2-5 During the secondary-progressive phase of the disease, inflammation and relapses occur infrequently, axonal loss is increased and disability progresses. MRI-defined plaque burden and clinical impairment accumulate over time. 1. Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359: Erratum in: Lancet. 2002;360:648. 2. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. Neurology. 1996;46: 3. Olerup O, Hillert J, Fredrikson S, et al. Primary chronic progressive and relapsing/remitting multiple sclerosis: two immunogenetically distinct disease entities. Proc Natl Acad Sci U S A. 1989;86: 4. Thompson AJ, Kermode AG, MacManus DG, et al. Patterns of disease activity in multiple sclerosis: clinical and magnetic resonance imaging study. BMJ. 1990;300: 5. Revesz T, Kidd D, Thompson AJ, Barnard RO, McDonald WI. A comparison of the pathology of primary and secondary progressive multiple sclerosis. Brain. 1994;117: Inflammation Axonal Loss Frequent inflammation, demyelination, axonal transection, plasticity, and remyelination Continuing inflammation, persistent demyelination Infrequent inflammation, chronic axonal degeneration, gliosis Compston A, Coles A. Lancet. 2002;359:

41
Aspettative di vita Sopravvivenza media > 25 anni
Morte per cause correlate alla disabilità ma indipendenti dalle lesioni neurologiche (broncopolminiti, polminiti ab ingestissepsi etc)
")
42
Prognosi Fattori prognostici positivi Fattori negativi
Esordio giovanile Sintomi monofocali Interessamento troncale e n.ottico Sintomi sensitivi Recupero totale >tempo intercorrente tra i primi due attacchi Fattori negativi Esordio tardivo Precoce interessamento cerebellare o piramidale Esordio multifocale

43
Indagini paracliniche
TC encefalo RMN Potenziali evocati (PEV, BAEP, SEP) Liquor (IgG index; Bande oligoclonali)
 Liquor (IgG index; Bande oligoclonali)")
44
Risonanza Magnetica Nucleare
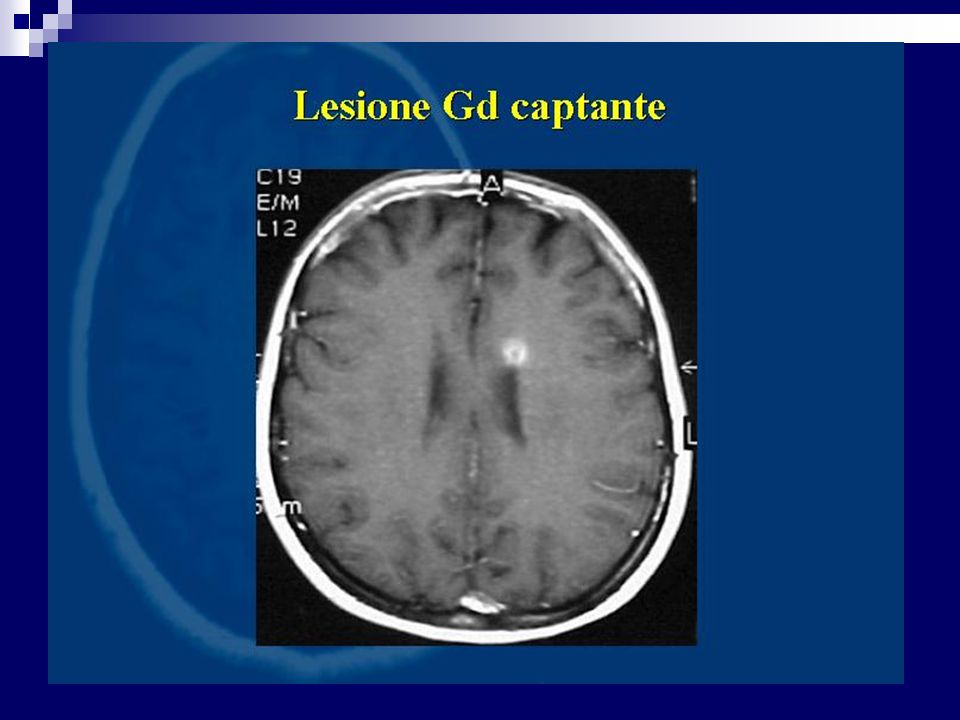
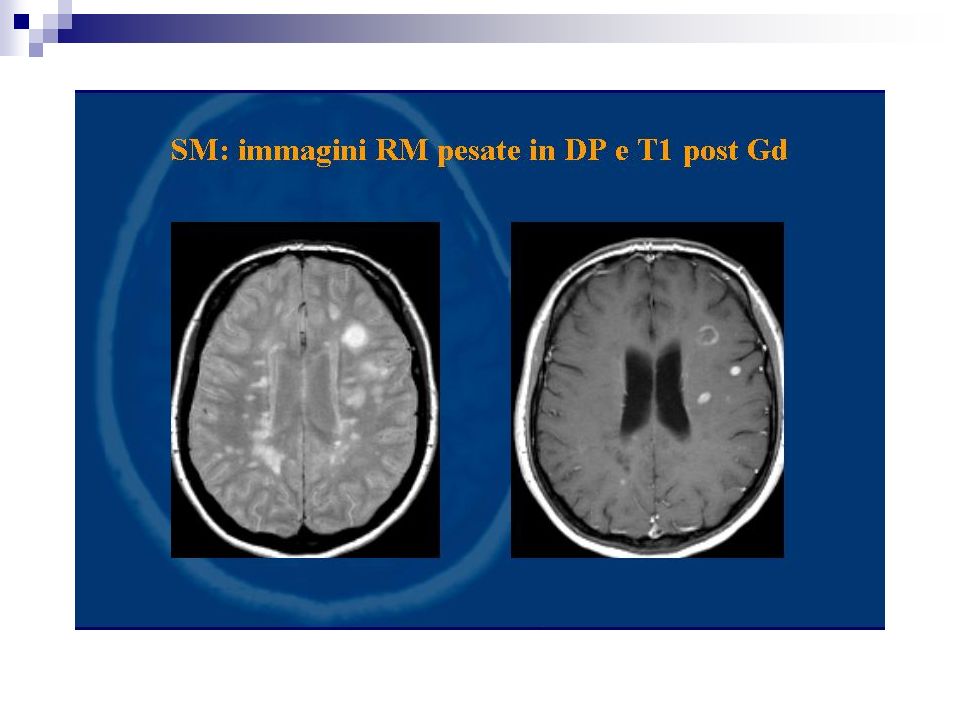
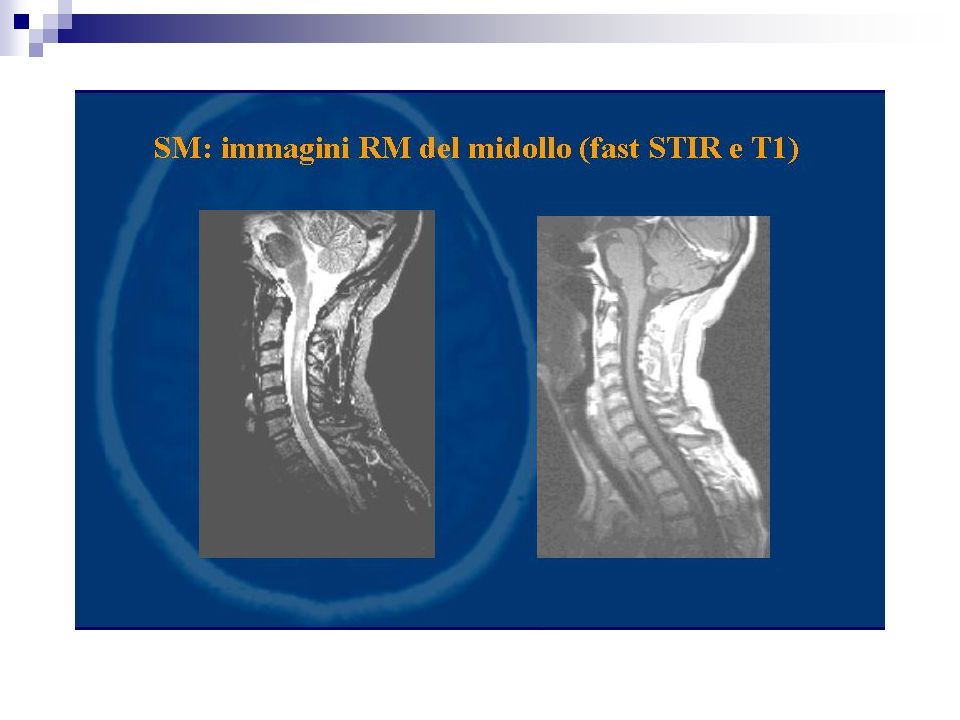
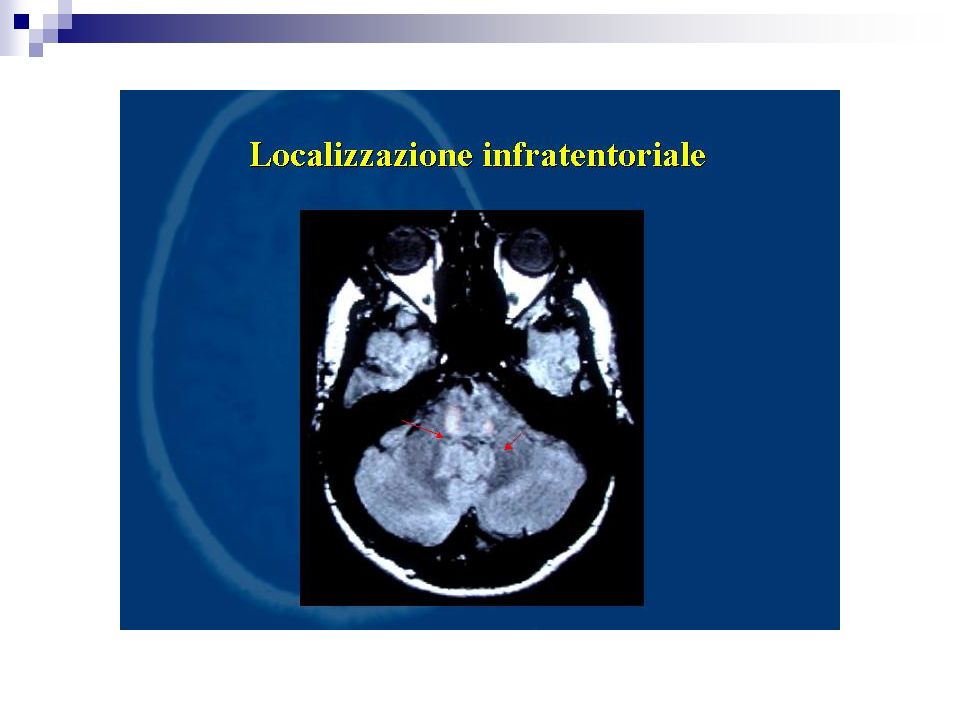
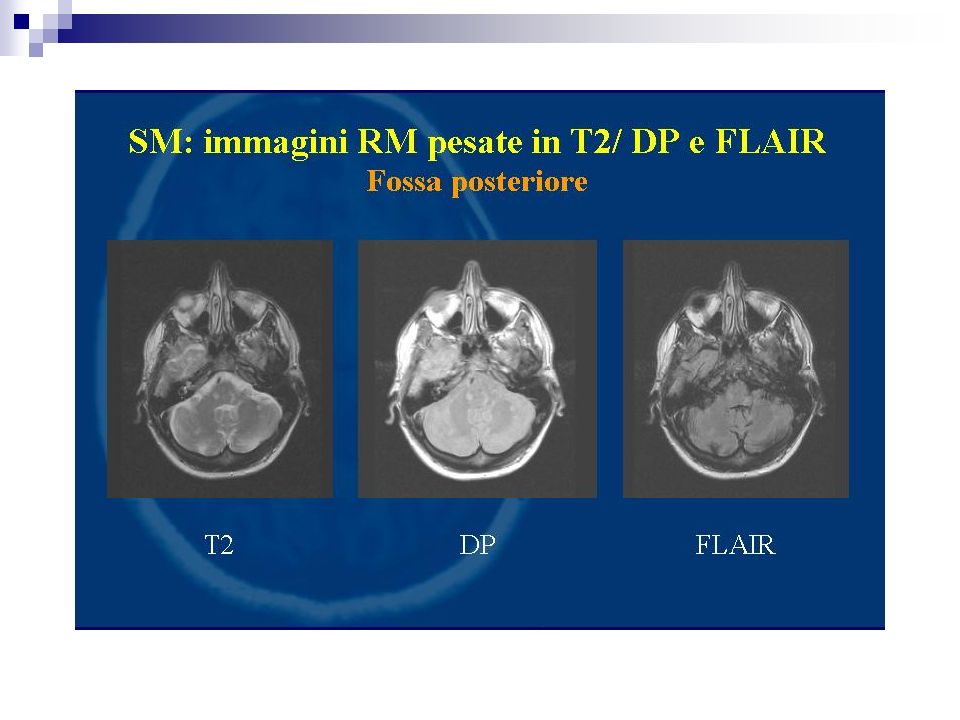
Percentuale di positività 90% dei casi definiti Più bassi i valori di specificità. I focolai di demielinizzazione appaiono iperintensi in T2 e DP prevalentemente disposti attorno i ventricoli laterali, possono confluire. Evidenziabili lesioni anche a livello del tronco e del midollo L’impiego di gadolinio (contrasto paramagnetico) fornisce elementi utili sulla dimostrazione di placche con rottura di barriera permettendo un giudizio sul grado di attività
 fornisce elementi utili sulla dimostrazione di placche con rottura di barriera permettendo un giudizio sul grado di attività.")
45
Potenziali evocati PEV BAEP PESS
Permette lo studio delle vie ottiche ed il riscontro di lesioni silenti dimostrando la disseminazione spaziale Alterati 60-80% casi (Aumento di latenza dell’onda P100) BAEP Utili nell’evidenziare lesioni tronco-encefaliche Alterati nel 40-60% di casi 40% casi asintomatici per lesioni del tronco encefalo Anomalie polimorfe ad eccezione dell’onda I PESS Esplorano l’integrità del midollo e del troncoencefalo Alterati solo nel 60% dei casi
 BAEP. Utili nell’evidenziare lesioni tronco-encefaliche. Alterati nel 40-60% di casi. 40% casi asintomatici per lesioni del tronco encefalo. Anomalie polimorfe ad eccezione dell’onda I. PESS. Esplorano l’integrità del midollo e del troncoencefalo. Alterati solo nel 60% dei casi.")
46
Alterazione del CSF Presenza di bande oligoclonali IgG 95%
Aumento dell’indice di Link 90% Aumento delle IgG/albumina 80% Aumento delle IgG/proteine tot 70% Aumento delle IgG 60% Modesta pleiocitosi 50% Modesta proteinorrachia 30%

47
Criteri di Poser (1983) SM clinicamente definita
SM clinicamente probabile SM definita con supporto di laboratorio SM probabile con supporto di laboratorio

48
Criteri di Mc Donald (2000) Obbiettivo primario: dimostrazione della disseminazione nel tempo e nello spazio delle lesioni.
 Obbiettivo primario: dimostrazione della disseminazione nel tempo e nello spazio delle lesioni.")
49
Criteri di Mc Donald (2000) PRESENTAZIONE CLINICA
2 o + attacchi; evidenza clinica obbiettiva di due o + lesioni 2 o + attacchi; evidenza clinica obbiettiva di 1 lesioni DATI AGGIUNTIVI NECESSARI Nessuno Disseminazione spaziale dimostrata alla RMN o 2 o più lesioni alla RM suggestive per SM e positività al LCR attendere un nuovo attacco coinvolgente una sede diversa

50
Criteri di Mc Donald (2000) PRESENTAZIONE CLINICA
1 attacco; evidenza clinica di 2 o più lesioni 1 attacco; evidenza clinica obiettiva di 1 lesione presentazione monosintomatica sindrome clinicamente isolata DATI AGGIUNTIVI NECESSARI Disseminazione temporale dimostrata con RM o secondo attacco clinico Disseminazione spaziale dimostrata alla RMN o 2 o più lesioni alla RM suggestive per SM e positività al LCR con disseminazione temporale dimostrata con RM o secondo attacco clinico

51
Criteri di Mc Donald (2000) PRESENTAZIONE CLINICA
DATI AGGIUNTIVI NECESSARI Liquor positivo e disseminazione dimostrata ad: a) 9 o + leioni in T2 b) 2 o + llesioni midollari o c) 4-8 lesioni cerebrali e 1 midollare d) ??? o PEV alterati associati a 4-8 lesioni cerebrali, o con meno di 4 lesioni più una lesione midollare alla RM e disseminazione temporale dimostrata alla RM o Progressione continua per 1 anno PRESENTAZIONE CLINICA Progressione neurologica insidiosa suggestiva per SM
 9 o + leioni in T2. b) 2 o + llesioni midollari o. c) 4-8 lesioni cerebrali e 1 midollare. d) o. PEV alterati associati a 4-8 lesioni cerebrali, o con meno di 4 lesioni più una lesione midollare alla RM e disseminazione temporale dimostrata alla RM o. Progressione continua per 1 anno. PRESENTAZIONE CLINICA. Progressione neurologica insidiosa suggestiva per SM.")
63
Histopathologic Correlates of Black Holes T1 Hypointensity Directly Correlates to Degree of Axonal Loss Bodian axonal density 1. Strongly hypointense 20% 2. Mildly hypointense 60% Histopathologic Correlates of Black Holes T1 Hypointensity Directly Correlates to Degree of Axonal Loss Combined histopathologic and MRI studies have provided the most compelling evidence that marked hypointense T1 lesions reflect severe tissue damage.1,2 These studies have demonstrated a strong correlation between lesion hypointensity on T1-weighted images and the percentage of residual axons. In the van Waesberghe study,1 the histopathology of postmortem tissue samples from 17 MS patients was evaluated on the basis of T2-weighted imaging, including NAWM and T1 hypointense lesions. In each sample, MTRs, T1 contrast ratio, myelin, and axonal density was measured. “Postmortem tissue sampling by MRI revealed a range of pathology, illustrating the high sensitivity and low specificity of T2-weighted imaging. T1 hypointensity and MTR were strongly associated with axonal density, emphasizing their role in monitoring progression in multiple sclerosis.”1 1. van Waesberghe JH, Kamphorst W, De Groot CJ, et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol. 1999;46: 2. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343: Other Studies Histopathology: severe myelin and axon loss. Brück W, Bitsch A, Kolenda H, Brück Y, Stiefel M, Lassmann H. Inflammatory central nervous system demyelination: correlation of magnetic resonance imaging findings with lesion pathology. Ann Neurol. 1997:42: van Waesberghe JH, Kamphorst W, De Groot CJ, et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insight into substrates of disability. Ann Neurol. 1999;46: MTI: markedly reduced MTR. van Walderveen MA, Barkhof F, Pouwels PJ, van Schijndel RA, Polman CH, Castelijns JA. Neuronal damage in T1-hypointense multiple sclerosis lesions demonstrated in vivo using proton magnetic resonance spectroscopy. Ann Neurol. 1999;46:79-87. MRS: markedly reduced NAA. Loevner LA, Grossman RI, McGowan JC, Ramer KN, Cohen JA. Characterization of multiple sclerosis plaques with T1-weighted MR and quantitative magnetization transfer. AJNR Am J Neuroradiol. 1995;16: DTI: markedly increased diffusivity and reduced fractional anisotropy. Filippi M, Cercignani M, Inglese M, Horsfield MA, Comi G. Diffusion tensor magnetic resonance imaging in multiple sclerosis. Neurology. 2001;56: van Waesberghe JH, van Walderveen MA, Castelijns JA, et al. Patterns of lesion development in multiple sclerosis: longitudinal observations with T1-weighted spin-echo and magnetization transfer MR. AJNR Am J Neuroradiol. 1998;19: 3. NAWM 100% Reprinted with permission from Noseworthy JH et al. N Engl J Med. 2000;343: Reprinted with permission from van Waesberghe JH et al. Ann Neurol. 1999;46:

64
Neuronal Dysfunction Viewed by MRI
Atrophy Control MS Patient Neuronal Dysfunction Viewed by MRI Atrophy MRI has made it possible to image the MS lesions during life and to monitor various aspects of the lesions as they evolve.1 Gross brain atrophy can now be detected with great accuracy. Atrophy and several other measures indicate that tissue loss occurs from the earliest stages of the disease and not simply from the time that detectable disability accumulates, which can be later in the disease course. Brain atrophy correlates quite closely with progression, whereas relapse frequency is only weakly related to lesion load, brain atrophy, and the progression of disability. 1. van Walderveen MA, Barkhof F, Pouwels PJ, van Schijndel RA, Polman CH, Castelijns JA. Neuronal damage in T1-hypointense multiple sclerosis lesions demonstrated in vivo using proton magnetic resonance spectroscopy. Ann Neurol. 1999;46:79-87. Adapted with permission from van Walderveen MA et al. Ann Neurol. 1999;46:79-87.

65
Pathogenic Pathways Potential Treatment Strategies
Intact CNS Anti-inflammation Axon Protection Inflammation Demyelination Axonal Damage Pathogenic Pathways Potential Treatment Strategies Carefully dissecting the pathogenic pathways of MS reveals inflammation and degeneration as two potential treatment targets.1 This diagram depicts the different areas where the current disease-modifying drugs, such as glatiramer acetate and interferon-, mediate their biologic effects. 1. Rieckmann P, Maürer M. Anti-inflammatory strategies to prevent axonal injury in multiple sclerosis. Curr Opin Neurol. 2002;15: Adapted with permission from Rieckmann P, Maürer M. Curr Opin Neurol. 2002;15:

66
Terapie patogenetiche (Immunosoppressori ed immunomodulatori)
Interferone beta umano ricombinante Copolimero-1 Azatioprina Methotrexate Ciclofosfamide Mitoxantrone

67
Terapie immunomodulanti
Interferone beta Immunoglobuline Copolimero-1 Terapie combinate

68
Interferone beta Avonex (IFNb-1a): 6MU (30mg) i.m. sett.
Rebif (IFNb-1a): 6MU (22mg) o 12 MU (44mg) s.c 3 volte sett. Betaferon (IFNb-1b): 8 MU s.c.a giorni alterni. Effetti collaterali: sindromepseud-influenzale, aumento degli enzimi epatici, depressione leucopenia, anticoerpi anti tiroide Efficacia: Riduzione del relapse rate ( ) lieve progressione di malattia all’EDSS
: 6MU (22mg) o 12 MU (44mg) s.c 3 volte sett. Betaferon (IFNb-1b): 8 MU s.c.a giorni alterni. Effetti collaterali: sindromepseud-influenzale, aumento degli enzimi epatici, depressione leucopenia, anticoerpi anti tiroide. Efficacia: Riduzione del relapse rate ( ) lieve progressione di malattia all’EDSS.")
69
Copolimero-1 o Glatiramer acetato
Sale acetato di una mistura di polipeptidi sintetici Somministrazione 20mg/die s.c. Efficacia: sovrapponibile all’IFN Effetti collaterali: Reazioni locali; crisi pseudo-anginose dopo la somministrazione che si risolvono spontaneamemte dopo circa 15 min.

70
Immunosoppressori Azatioprina: 2.5 mg/kg/die
Ciclofosfamoide: 1-2g/m2 di superficie corporea mese Mitoxantrone: 20mg al mese associato ad 1 g ev di metilprednisolone Methotrexate: mg settimana Irradiazione

71
Trattamento delle ricadute
Bolo steroideo MP 1 gr diluito in 500 ml di fisiologica in 1-2 ore per 5 gg consecutivi associato ad un gastroprotettore Progressiva riduzione nei successivi 10 gg

72
Terapie sintomatiche Bolo steroideo Terapia per la spasticità
Terapia della fatica Terapia del tremore Fenomeni parossistici Disturbi sfinterici

73
Terapie sintomatiche Terapia per la spasticità Terapia della fatica
(Baclofene, Dantrolene, Tizanidina, Gabapentim, Benzodiazepine) Terapia della fatica (4-aminopiridina, amantadina, Pemolina) Terapia del tremore (Benzodiazepine, Valproato di sodio, primidone, propanololo) Fenomeni parossistici Baclofene, CBZ, Difenilidantoina Gabapentim)
 Terapia della fatica. (4-aminopiridina, amantadina, Pemolina) Terapia del tremore. (Benzodiazepine, Valproato di sodio, primidone, propanololo) Fenomeni parossistici. Baclofene, CBZ, Difenilidantoina Gabapentim)")
74
Terapie sintomatiche Disturbi della minzione: Disturbi sessuali
- Alterazione del riempimento - Alterazione dello svuotamento - Combinazione dei due tipi Disturbi sessuali Disturbi intestinali

75
Malattie demielinizzanti
Malattia di Schilder Diffusa demielinizzazione e gliosi dei centri semiovali. Forme infantili co disturbi visivi, psichici, convulsività e s.i.e. Neuromielite ottica acuta di Devic Sclerosi concentrica di von Balò Aree concentriche di demielinizzazione Sclerosi Multipla fulminante tipo Marburg

77
Sclerosi Multipla Patologia infiammatoria demielinizzante, multifocale sia temporalmente sia spazialmente del sistema nervoso centrale a verosimile patogenesi autoimmune

82
Malattie dismielinizzanti Leucodistrofie
Geneticamente determinate Alterazione della sintesi o del metabolismo della mielina Alterazioni simmetriche e diffuse della SB Decorso progressivo e multifocale deterioramento mentale progressivo ritardo o arresto sviluppo psicomotorio

83
Classificazione LD a difetto metabolico noto
Malattia Ereditarietà Deficit metabolico M. Di Krabbe AR Galattosilceramide-b galattosidasi LD metacromatica AR Arilsulfatasi A Adrenoleucodistrofia R X-Link. Enz. perossisomiali M. Di Zellweger AR Enz. perossisomiali M. Di Canavan AR Aspartato ciclasi M. Di Pelizaeus- X-Link Prot. proteolipidica Merzbacher

Presentazioni simili

















 SPECIFICA/SELETTIVA:>")






>")

>")

