Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
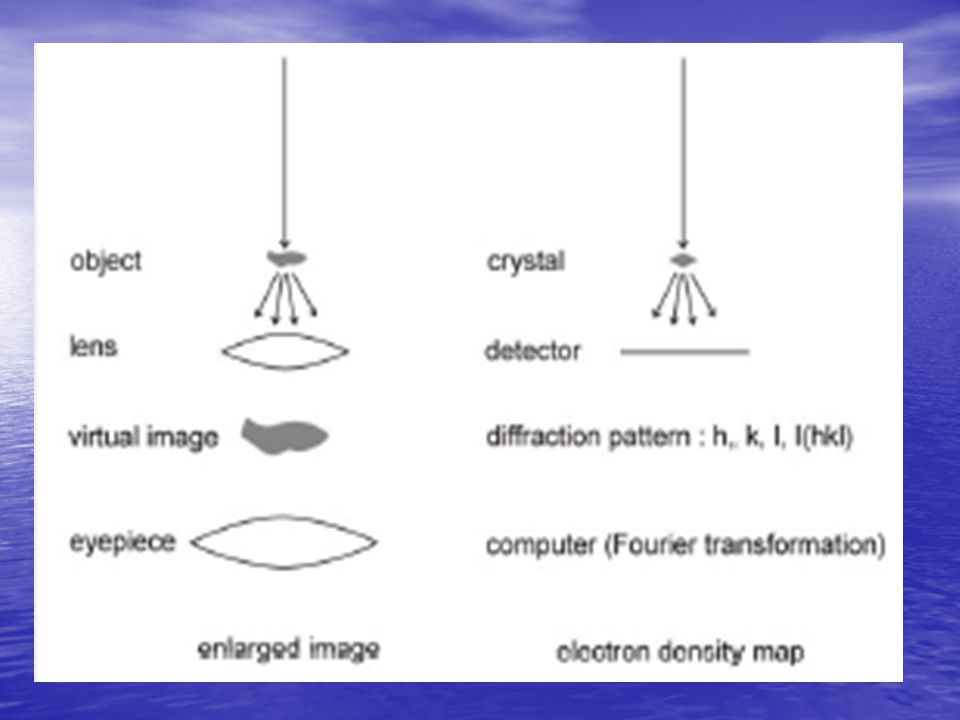
Determinazione della fase
Cristallografia delle piccole molecole Metodi diretti Metodo della Sostituzione Isomorfa Molecole Grandi (Proteine) (Sostituzione Isomorfa Multipla – MIR, multiple isomorphous replacement). MIR – Introduzione di nuovi centri di scattering nella cella elementare: costituiti da atomi pesanti (in modo che essi contribuiscano significativamente alla diffrazione) in numero limitato (per localizzarne la posizione) non dovrebbero cambiare le caratteristiche strutturali della molecola e della cella (cristalli isomorfi). In pratica la MIR si ottiene facendo diffondere diversi complessi di metalli pesanti nei canali dei cristalli preformati. Se le catene espongono gruppi –SH, essi legheranno i metalli. Nelle metallo-proteine i metalli leggeri possono essere sostituiti con metalli più pesanti (es. Zn-Hg, Ca-Sm).
 (Sostituzione Isomorfa Multipla – MIR, multiple isomorphous replacement). MIR – Introduzione di nuovi centri di scattering nella cella elementare: costituiti da atomi pesanti (in modo che essi contribuiscano significativamente alla diffrazione) in numero limitato (per localizzarne la posizione) non dovrebbero cambiare le caratteristiche strutturali della molecola e della cella (cristalli isomorfi). In pratica la MIR si ottiene facendo diffondere diversi complessi di metalli pesanti nei canali dei cristalli preformati. Se le catene espongono gruppi –SH, essi legheranno i metalli. Nelle metallo-proteine i metalli leggeri possono essere sostituiti con metalli più pesanti (es. Zn-Hg, Ca-Sm).")
2
Determinazione della fase
I metalli pesanti contengono molti più elettroni degli atomi leggeri (H,N,O,C,S) della proteina, diffondono i RX più intensamente. Dopo una MIR se tutte le interferenze fossero positive tutti i raggi diffratti aumenterebbero di intensità. Ma alcune interferenze sono negative, per cui dopo la MIR, alcuni spot aumentano di intensità, altri diminuiscono ed altri non variano. Dalle differenze di intensità prima e dopo la MIR, si possono dedurre le posizioni degli atomi pesanti nel cristallo. Le trasformate di Fourier di queste diff. di intensità forniscono le mappe dei vettori tra gli atomi pesanti (mappe di Patterson). Dalle mappe di Patterson si possono dedurre le posizioni degli atomi pesanti nella cella elementare, per cui da queste le ampiezze e le fasi e dal loro contributo ai raggi diffratti dal cristallo.
 della proteina, diffondono i RX più intensamente. Dopo una MIR se tutte le interferenze fossero positive tutti i raggi diffratti aumenterebbero di intensità. Ma alcune interferenze sono negative, per cui dopo la MIR, alcuni spot aumentano di intensità, altri diminuiscono ed altri non variano. Dalle differenze di intensità prima e dopo la MIR, si possono dedurre le posizioni degli atomi pesanti nel cristallo. Le trasformate di Fourier di queste diff. di intensità forniscono le mappe dei vettori tra gli atomi pesanti (mappe di Patterson). Dalle mappe di Patterson si possono dedurre le posizioni degli atomi pesanti nella cella elementare, per cui da queste le ampiezze e le fasi e dal loro contributo ai raggi diffratti dal cristallo.")
3
Determinazione della fase
Conoscendo: Ampiezza e fase atomi pesanti Ampiezza proteina Ampiezza proteina-metalli pesanti si può calcolare se l’interferenza dei RX diffusi dai metalli pesanti e dalla proteina è costruttiva o distruttiva. Una stima della fase della proteina si può ottenere dall’entità dell’interferenza con le informazioni sulla fase del metallo. Sono però possibili due differenti angoli di fase, per distinguere tra le soluzioni possibili è necessario avere un complesso con un secondo atomo pesante che darà altri due angoli di fase possibili per la proteina. Quello che avrà lo stesso valore di uno degli angoli di fase determinati nel caso precedente sarà l’angolo di fase corretto. In pratica si devono preparare più di due complessi proteina-metallo pesante per ottenere una fase buona per tutte le riflessioni. Le ampiezze e le fasi dei dati ottenuti vengono impiegati per calcolare le mappe di densità elettronica dell’unità ripetitiva del cristallo.

5
Costruzione del modello
La mappa di densità elettronica va interpretata in termini di catena peptidica (sequenza aa). Vi sono però limiti dei dati: imprecisioni da errori negli angoli di fase risoluzione dei dati che dipende dall’ordine nei cristalli (misurata in A, minore è il suo valore più elevata è la risoluzione e quindi la quantità di dettagli che si possono osservare). Bassa risoluzione (>5) forma della molecola, regioni a elica come cilindri Media risoluzione (ca.3) percorso della catena e fitting sequenza aa Alta risoluzione ca.2 – si discrimina tra catene laterali molto simili ca.1 – atomi come sfere di densità elettronica La costruzione del modello iniziale è un processo di prova/errore. Occorre: decidere in che modo la catena polipetidica traccia il suo percorso nella densità elettronica si tenta di adattare in base all’ipotesi la densità delle catene laterali della sequenza nota del polipetide Utilizzo computer grafica
. Vi sono però limiti dei dati: imprecisioni da errori negli angoli di fase. risoluzione dei dati che dipende dall’ordine nei cristalli (misurata in A, minore è il suo valore più elevata è la risoluzione e quindi la quantità di dettagli che si possono osservare). Bassa risoluzione (>5) forma della molecola, regioni a elica come cilindri. Media risoluzione (ca.3) percorso della catena e fitting sequenza aa. Alta risoluzione ca.2 – si discrimina tra catene laterali molto simili ca.1 – atomi come sfere di densità elettronica. La costruzione del modello iniziale è un processo di prova/errore. Occorre: decidere in che modo la catena polipetidica traccia il suo percorso nella densità elettronica. si tenta di adattare in base all’ipotesi la densità delle catene laterali della sequenza nota del polipetide. Utilizzo computer grafica.")
6
Costruzione del modello
La mappa di densità elettronica viene rappresentata sullo schermo del computer (computer grafica) in combinazione con una parte della catena polipeptidica. La mappa di densità elettronica ottenuta viene interpretata adattandovi parti della catena polipeptidica con stechiometria nota (backbone e catene laterali). Le unità della catena polipeptidica vengono ruotate e traslate rispetto alla densità elettronica fino a che si ottiene una buona corrispondenza tra le due. Utilizzo mappa Fourier differenza (sia per ‘aggiustare’ modello che per risolvere nuove strutture).
 in combinazione con una parte della catena polipeptidica. La mappa di densità elettronica ottenuta viene interpretata adattandovi parti della catena polipeptidica con stechiometria nota (backbone e catene laterali). Le unità della catena polipeptidica vengono ruotate e traslate rispetto alla densità elettronica fino a che si ottiene una buona corrispondenza tra le due. Utilizzo mappa Fourier differenza (sia per ‘aggiustare’ modello che per risolvere nuove strutture).")
7
Costruzione del modello – R-factor
Il modello viene via via corretto mediante affinamento, per cui esso viene modificato minimizzando la differenza tra ampiezze osservate sperimentalmente e quelle calcolate per un ipotetico cristallo contenente il modello invece della molecola reale (minimi quadrati). Questa differenza viene espressa come R-factor che rappresenta la discordanza residua (R=0 per un accordo esatto, 0.59 per disaccordo totale). Per una struttura proteica ben determinata R è compreso tra 0.15 e 0.20. La diff. residua (ad alta risoluzione) non è dovuta a grossi errori nel modello, ma a errori e inesattezze nei dati che derivano principalmente da: Piccole variazioni nelle conformazione delle molecole proteiche Correzioni inadeguate presenza solvente Differenze orientamento microcristalli che formano il cristallo. Il modello finale è media delle molecole (leggermente diverse in conformazione e orientamento) presenti nel cristallo, per cui il modello non corrisponde mai in modo esatto al cristallo reale. La conoscenza della sequenza aa (da sequenza nucleotica nelle tecniche DNA ricmbinante per la produzione della proteina stessa) è indispensabile per la determinazione della struttura a RX. Le strutture determinate possono identificare errori nella determinazione della sequenza.
. Questa differenza viene espressa come R-factor che rappresenta la discordanza residua (R=0 per un accordo esatto, 0.59 per disaccordo totale). Per una struttura proteica ben determinata R è compreso tra 0.15 e La diff. residua (ad alta risoluzione) non è dovuta a grossi errori nel modello, ma a errori e inesattezze nei dati che derivano principalmente da: Piccole variazioni nelle conformazione delle molecole proteiche. Correzioni inadeguate presenza solvente. Differenze orientamento microcristalli che formano il cristallo. Il modello finale è media delle molecole (leggermente diverse in conformazione e orientamento) presenti nel cristallo, per cui il modello non corrisponde mai in modo esatto al cristallo reale. La conoscenza della sequenza aa (da sequenza nucleotica nelle tecniche DNA ricmbinante per la produzione della proteina stessa) è indispensabile per la determinazione della struttura a RX. Le strutture determinate possono identificare errori nella determinazione della sequenza.")
8
Determinazione struttura proteine
Cristallo Densità elettronica r (x,y,z) Data Collection |F|, (hkl) Fasi a(hkl) Struttura
 Data Collection. |F|, (hkl) Fasi a(hkl) Struttura.")
9
GMER

10
Actina

Presentazioni simili
>")





![PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]](/1/540371/big_thumb.jpg "PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]>")
>")











>")