Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Esempio di importanza della farmacocinetica:
MORFINA ED EROINA farmaco Assorbimento, Distribuzione, metabolismo Metabolismo, Escrezione,etc cinetica Farmaco attivo risposta R dinamica

2
Specifico? Niente e’ specifico
[amitriptilina] -8 -7 -6 -5 -4 -3 -2 Antagonista H1 Inibitore ricaptazione catecolamine |antagonista H2 Antagonista a1 Antagonista muscarinico Inib. fosfodiesterasi

3
Dose massima tollerata
Farmacocinetica tossicita’ effetti collaterali [farmaco] efficace inefficace 1 2 3 4 5 6 7 8 9 10 tempo Indici terapeutici Dose massima tollerata Dose minima efficace Dose letale 50 Dose efficace 50

4
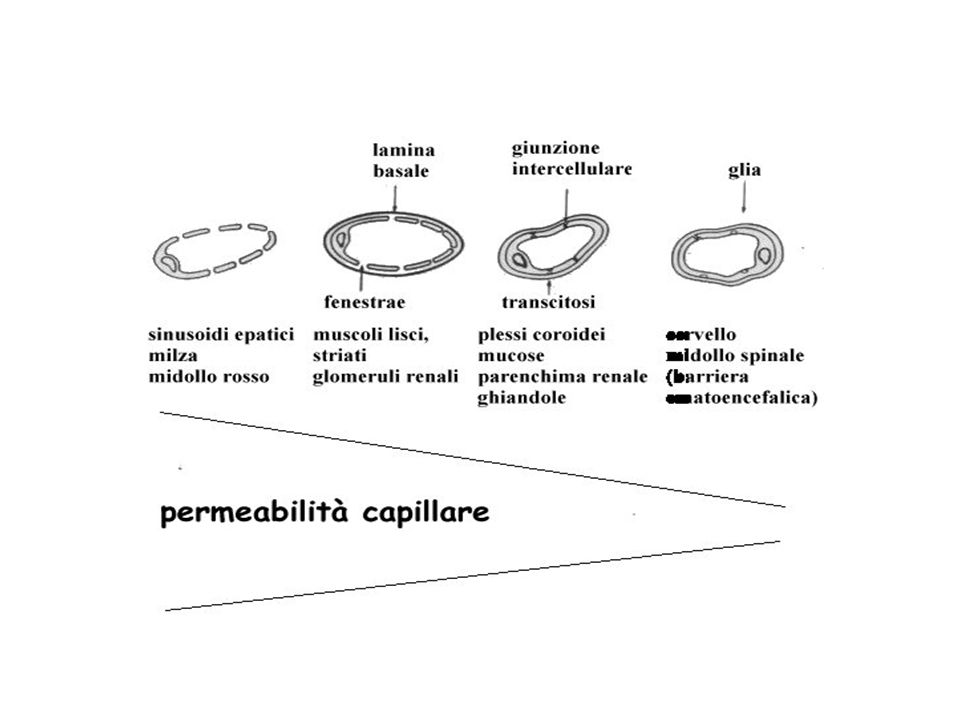
Torrente circolatorio
Farmaco Torrente circolatorio Recettore bersaglio Cellule bersaglio Concentrazione sufficiente Degradazione ed escrezione

5
farmacocinetica

6
TIPI DI SOMMINISTRAZIONI IN FARMACOLOGIA
Somministrazione enterale Orale Sublinguale Rettale Somministrazione parenterale Intravenosa intramuscolare Sottocutanea Somministrazione polmonare aerosol Applicazione topica

7
3 meccanismi principali: - Diffusione passiva
Assorbimento 3 meccanismi principali: - Diffusione passiva - Diffusione facilitata (e.g. levodopa) - Trasporto attivo (e.g. 5-fluorouracile) anche cianuro, paraquat, DDT anche inverso - proteine MDR
 - Trasporto attivo (e.g. 5-fluorouracile) anche cianuro, paraquat, DDT. anche inverso - proteine MDR.")
8
Farmaco Lipidi Soluz. acquosa Soluz. Acquosa Diffusione passiva
Legge di Fick Vd = k a (c2 - c1) Lipidi Soluz. acquosa Soluz. Acquosa Farmaco Grandezza della molecola Spessore della membrana Concentrazione del farmaco Superficie della membrana Preparazione farmaceutica Vascolarizzazione Ionizzazione Tempo Lipofilia
 Lipidi. Soluz. acquosa. Soluz. Acquosa. Farmaco. Grandezza della molecola Spessore della membrana. Concentrazione del farmaco Superficie della membrana. Preparazione farmaceutica Vascolarizzazione. Ionizzazione Tempo. Lipofilia.")
9
[non-ionizzata] [ionizzata] [ionizzata] [non-ionizzata]
La pKa determina la tendenza di una molecola a trovarsi in forma ionizzata Equazione di Henderson - Hasselbach [non-ionizzata] [ionizzata] Per gli acidi: pKa - pH = log [ionizzata] [non-ionizzata] Per le basi: pKa - pH = log
![[non-ionizzata] [ionizzata] [ionizzata] [non-ionizzata]](http://slideplayer.it/slide/596600/2/images/9/%5Bnon-ionizzata%5D+%5Bionizzata%5D+%5Bionizzata%5D+%5Bnon-ionizzata%5D.jpg "La pKa determina la tendenza di una molecola a trovarsi in forma ionizzata. Equazione di Henderson - Hasselbach. [non-ionizzata] [ionizzata] Per gli acidi: pKa - pH = log. [ionizzata] [non-ionizzata] Per le basi: pKa - pH = log.")
10
pKa: misura della forza di interazione di una molecola con un protone
Quando il pKa di un farmaco è uguale al pH del liquido circostante, saranno presenti in uguale misura molecole ionizzate e non ionizzate. Se il pKa è uguale al pH a cui avviene il 50% della ionizzazione. Se un farmaco ha pKa di 4, vuol dire che a pH 4 il 50% del farmaco sarà ionizzato etc. pKa: misura della forza di interazione di una molecola con un protone pKa = acido forte pKa = base forte

11
pH basso Acido ionizzato Acido non ionizzato pH alto pH alto Base ionizzata Base non ionizzata pH basso Non permeabile permeabile

12
Il naprossene come esempio (pKa = 5)
pH 1.4 pH 7.4 [1] HA HA [1] [.001] A- [252] pH alto Acido ionizzato acido non ionizzato pH basso Succo gastrico Mucosa gastrica plasma Per acidi deboli: maggiore il pH, maggiore la ionizzazione

13
L’intestino e’ il sito principale per l’assorbimento

14
La morfina come esempio (pKa = 8)
pH 5.3 pH 7.4 [1] BA BA [1] [501] B+ [4] pH alto Base ionizzata Base non ionizzata pH basso intestino Mucosa plasma Per basi deboli: maggiore il pH, maggiore l’assorbimento

15
COEFFICIENTE DI RIPARTIZIONE
E’ molto importante la solubilità del farmaco nel doppio strato lipidico, dato dal che indica come un farmaco si distribuisce in una soluzione contenente H2O e olio (o n-ottanolo): Se > il farmaco è lipofilo e diffonde facilmente Se < il farmaco è idrofilo e non diffonde facilmente COEFFICIENTE DI RIPARTIZIONE COEFFICIENTE DI [farmaco] nella fase oleosa = RIPARTIZIONE [farmaco] nella fase acquosa Il coefficiente di ripartizione non è un parametro fisso, ma può variare in diverse situazioni, per esempio: la maggior parte dei farmaci sono acidi o basi deboli, quindi il coefficiente varia a seconda del pH dell’ambiente nel quale si trovano (questa variabile può essere sfruttata anche per aumentare la velocità di eliminazione: alcalinizzazione delle urine in caso di avvelenamento da barbiturici)
: Se > 1 il farmaco è lipofilo e diffonde facilmente. Se < 1 il farmaco è idrofilo e non diffonde facilmente. COEFFICIENTE DI RIPARTIZIONE. COEFFICIENTE DI [farmaco] nella fase oleosa. = RIPARTIZIONE [farmaco] nella fase acquosa. Il coefficiente di ripartizione non è un parametro fisso, ma può variare in diverse situazioni, per esempio: la maggior parte dei farmaci sono acidi o basi deboli, quindi il coefficiente varia a seconda del pH dell’ambiente nel quale si trovano (questa variabile può essere sfruttata anche per aumentare la velocità di eliminazione: alcalinizzazione delle urine in caso di avvelenamento da barbiturici)")
16
Ionizzazione e coefficiente di ripartizione non sono identici
Esempio: Barbitale (pKa = 7.8; coefficiente di ripartizione 1) assorbimento: 5% della dose dallo stomaco in 1 ora Secobarbitale (pKa = 7.9; coeffiente di ripartizione 52) assorbimento: 30% della dose dallo stomaco in 1 ora Tiopentale (pKa 7.6; coefficiente di ripartizione 580) assorbimento: 50% della dose dallo stomaco in 1 ora Mannitolo: (coefficiente di ripartizione 0.02) assorbimento: 1% o meno
 assorbimento: 5% della dose dallo stomaco in 1 ora. Secobarbitale (pKa = 7.9; coeffiente di ripartizione 52) assorbimento: 30% della dose dallo stomaco in 1 ora. Tiopentale (pKa 7.6; coefficiente di ripartizione 580) assorbimento: 50% della dose dallo stomaco in 1 ora. Mannitolo: (coefficiente di ripartizione 0.02) assorbimento: 1% o meno.")
17
CINETICHE DI ASSORBIMENTO
CINETICA DI I ORDINE: LA QUANTITA’ DI FARMACO ASSORBITA NELL’UNITA’ DI TEMPO E’ UNA % COSTANTE DI QUELLA CHE RIMANE DA ASSORBIRE La maggior parte dei farmaci diffonde per diffusione passiva, seguendo una cinetica di I ordine CINETICA DI ORDINE 0: LA QUANTITA’ DI FARMACO ASSORBITA NELL’UNITA’ DI TEMPO E’ COSTANTE Per alcuni farmaci l’assorbimento avviene attraverso meccanismi di trasporto attivo saturabili (la velocità di assorbimento dipende dal numero di trasportatori), seguendo una cinetica di ordine 0
, seguendo una cinetica di ordine 0.")
18
Effetto di primo passaggio
Farmaco Lume intestinale Cellule intestinali Fegato Polmone

19
Proprietà chimiche natura chimica
PROPRIETA’ CHIMICHE E VARIABILI FISIOLOGICHE IMPORTANTI CHE INFLUENZANO L’ASSORBIMENTO DI UN FARMACO Proprietà chimiche natura chimica peso molecolare solubilità coefficiente di ripartizione Variabili fisiologiche mobilità gastrica presenza di cibo nello stomaco pH nel sito di assorbimento area della superficie assorbente flusso ematico eliminazione presistemica

20
VIE DI SOMMINISTRAZIONE ENTERALI
corretta assunzione del farmaco Aumentato rischio di effetti collaterali utilizzata in emergenza evita l’effetto di primo passaggio assorbimento rapido l’effetto compare dopo pochi minuti SUBLINGUALE parziale effetto di primo passaggio ha una latenza d’azione minore rispetto alla via per os assorbimento variabile e incompleto RETTALE il pz deve essere sveglio e collaborante l’assorbimento incompleto può non permettere il raggiungimento della concentrazione minima efficace effetto di primo passaggio è la via più economica e più sicura possibilità di utilizzo di PREPARAZIONI RETARD assorbimento variabile, che dipende da molti fattori gli effetti compaiono dopo almeno minuti PER OS

21
VIE DI SOMMINISTRAZIONE PARENTERALI
non utilizzabile per grossi volumi dolore o necrosi (rara) utilizzando sostanze irritanti è utilizzata per soluzioni insolubili e per l’impianto di pellet solidi assorbimento rapido per le soluzioni acquose lento e prolungato per le preparazioni a lento rilascio SOTTOCUTANEA non utilizzabile se il pz. è in terapia con anticoagulanti si possono utilizzare volumi moderati si utilizza per somministrare sostanze oleose lento e prolungato per le preparazioni a lento rilascio INTRAMUSCOLO aumentato rischio di effetti collaterali l’infusione deve essere lenta non utilizzabile per sostanze oleose o insolubili utilizzata in emergenza possono essere iniettati grossi volumi si possono somministrare sostanze irritanti diluite (KCl) 100% assorbimento effetti immediati ENDOVENA Altre vie parenterali sono definite: VIE D’ORGANO
 utilizzando sostanze irritanti. è utilizzata per soluzioni. insolubili e per l’impianto di. pellet solidi. assorbimento rapido per le. soluzioni acquose. lento e prolungato per. le preparazioni a lento rilascio. SOTTOCUTANEA. non utilizzabile se il pz. è in terapia con anticoagulanti. si possono utilizzare volumi. moderati. si utilizza per somministrare. sostanze oleose. lento e prolungato per le. preparazioni a lento rilascio. INTRAMUSCOLO. aumentato rischio di effetti collaterali. l’infusione deve essere lenta. non utilizzabile per sostanze oleose o insolubili. utilizzata in emergenza. possono essere iniettati grossi volumi. si possono somministrare. sostanze irritanti diluite (KCl) 100% assorbimento. effetti immediati. ENDOVENA. Altre vie parenterali sono definite: VIE D’ORGANO.")
22
BIODISPONIBILITA’: e’ la frazione del farmaco/tossico somministratoche raggiunge la circolazione sistemica in forma immodificata. Si determina confrontando i livelli plasmatici di un farmaco somministrato per una certa via con quelli che si ottengono dopo somministrazione per endovena per la quale l’entita’ di assorbimento e’ del 100%. Metabolismo epatico Solubilita’ del farmaco Instabilita’ chimica Forma farmaceutica

23
Torrente circolatorio
Farmaco Torrente circolatorio Recettore bersaglio Cellule bersaglio Concentrazione sufficiente Degradazione ed escezione

24
Capillari

25
Perfusione relativa degli organi
Distribuzione Perfusione relativa degli organi totale ml min-1 100g-1 Polmoni Rene Fegato Intestino Cervello Muscolo scheletrico Grasso

26
Volume di distribuzione
= (quantita’ di farmaco nel corpo)/(concentrazione plasmatica) uomo di 70 Kg L/Kg sangue litri 0.1 extracellulare 12 litri 0.2 Liquido totale 42 litri 0.6 Eparina 0.1 L/Kg ? Warfarin 0.1 L/Kg ? Gentamicina 0.2 L/Kg Fenitoina 0.6 L/Kg Etanolo 0.6 L/Kg Imipramina 23 L/Kg ?
/(concentrazione plasmatica) uomo di 70 Kg. L/Kg. sangue 5.5 litri 0.1. extracellulare 12 litri 0.2. Liquido totale 42 litri 0.6. Eparina 0.1 L/Kg Warfarin 0.1 L/Kg Gentamicina 0.2 L/Kg. Fenitoina 0.6 L/Kg. Etanolo 0.6 L/Kg. Imipramina 23 L/Kg")
27
100 mg /1 L (100 mg/L) 100 mg /1 L+ 1 L (50 mg/L)
VOLUME DI DISTRIBUZIONE 100 mg 100 mg /1 L (100 mg/L) 100 mg /1 L+ 1 L (50 mg/L) E’ il rapporto tra la dose somministrata e la concentrazione massima risultante raggiunta nel plasma = (quantita’ di farmaco nel corpo)/(concentrazione plasmatica)
 100 mg /1 L+ 1 L. (50 mg/L) E’ il rapporto tra la dose somministrata e la concentrazione massima risultante. raggiunta nel plasma. = (quantita’ di farmaco nel corpo)/(concentrazione plasmatica)")
28
Proteine plasmatiche (e.g. albumina) warfarin
 warfarin")
29
Interazione tra farmaci
(e.g. albumina) 90% 80% Nuovo farmaco somministrato 10% 20% Warfarin Ibuprofen Fenitoina aspirina Furosemide
 90% 80% Nuovo farmaco. somministrato. 10% 20% Warfarin. Ibuprofen. Fenitoina. aspirina. Furosemide.")
30
Volume di distribuzione alto
Accumulo in specifici tessuti grasso cervello Tiopentale: sangue Altri tessuti cervello ipnosi Tessuto adiposo

32
Placenta: meno impermeabile del cervello

34
Torrente circolatorio
Farmaco Torrente circolatorio Recettore bersaglio Cellule bersaglio Concentrazione sufficiente Degradazione ed escezione

35
Metabolismo Puo’ portare a: Perdita di attivita’ Diminuzione di attivita’ Aumento di attivita’ Diversa attivita’ Attivazione (profarmaco - farmaco) Farmaci lipofilici sono piu’ difficilmente eliminabili (via urinaria, fecale e biliare).
 Farmaci lipofilici sono piu’ difficilmente eliminabili. (via urinaria, fecale e biliare).")
36
Principalmente nel fegato, ma anche nei polmoni, nella
Metabolismo Principalmente nel fegato, ma anche nei polmoni, nella parete intestinale o nel plasma Fegato Fase funzionalizzazione Fase coniugazione OH O - SO3- 1 2

37
Reazioni metaboliche di Fase 1 e 2
Assorbimento Metabolismo escrezione Fase 1 Fase 2 Metabolita con Attivita’ modificata coniugato Farmaco Metabolita inattivo coniugato Lipofilico Idrofilico

38
Contributo relativo di vari enzimi al metabolismo di fase 1 e 2
Glucuronato Solfato Metilico Acetato glutatione Citocromo P450 (e.g. ossidazione idrossilazione) Anche enzimi specifici: alcol-deidrogenasi
 Anche enzimi specifici: alcol-deidrogenasi.")
39
Citocromo P450 Uomo: 18 famiglie di CYP450, con 42 sottofamiglie e 57 geni Arabidopsis:249 CYP Riso: 324 Substrati endogeni: acidi grassi, steroidi, acidi biliari, vitamina D, Classificazione: CYP1A1 Substrati esogeni: sopratutto CYP1, CYP2, CYP3. classe sottoclasse

40
Si ricorderà che la farmacocinetica si suddivide in 4 principali fasi: l’assorbimento, la distribuzione, il metabolismo e l’escrezione. Malgrado il principale livello di interazione tra farmaci nella clinica sia il metabolismo, è importante ricordare che un farmaco può modificare l’efficacia di un altro farmaco in tutte le fasi farmacocinetiche. Facciamo un esempio: se ad un paziente viene somministrato un farmaco che riduce la motilità intestinale, altre terapie concomitanti potranno ovviamente subirne delle conseguenze. Sempre a livello di assorbimento, farmaci che sono in grado di influenzare il pH del tratto gastrointestinale modificano l’assorbimento di un alto numero di farmaci modificandone sia il livello di ionizzazione che la solubilità. Un altro tipo di interazione importante può verificarsi a livello della distribuzione delle proteine plasmatiche. La maggior parte dei farmaci è attiva nella sua forma libera, cioè non legata alle proteine. Se un paziente assume due farmaci che si legano alla stessa proteina, la disponibilità di farmaco libero, soprattutto del farmaco che mostra una affinità minore per questa proteina, sarà ovviamente aumentata. L’azione di farmaci come il warfarin, ad esempio, che si legano molto intensamente alle proteine plasmatiche, è estremamente sensibile a questo tipo di interazione. Un altro esempio lampante in questo senso è costituito dal probenecid o l’alcalinizzazione delle urine. Abbiamo ovviamente citato casi limite, ma è ovvio che molti dei farmaci correntemente in uso interagiscono in una maniera o in un’altra con altri farmaci. Chiaramente, non sono state prese in considerazione le interazioni a livello di meccanismo d’azione (farmacodinamiche), che possono manifestarsi sia con l’antagonismo, e quindi una riduzione della risposta terapeutica, che con il sinergismo, con amplificazione della risposta terapeutica e, a volte ma non sempre, degli effetti collaterali. Come conseguenza di quanto detto, in sintesi, si può ragionevolmente affermare: (1) la politerapia non deve mai essere trattata con sufficienza, sopratutto se le combinazioni dei farmaci sono nuove; (2) a parità di efficacia clinica sarebbe preferibile utilizzare farmaci che presentano meno interazioni con altri farmaci. In effetti, è importante ricordare che una totale mancanza di interazione tra farmaci è rara. Infatti, l’interazione farmacocinetica tra farmaci può anche non essere diretta. Mettiamo il caso di una co-somministrazione di antibiotici e anticoagulanti orali. La flora batterica intestinale, in parte responsabile della produzione di vitamina K, risulta diminuita dal trattamento antibiotico, per cui l’anticoagulante, che per essere assorbito compete con la vitamina K, sarà assorbito in misura maggiore (oltre a poter anche esplicare una maggiore attività in assenza della vitamina). Il quadro globale, infine, è ulteriormente complicato dalla necessità dell’organismo di metabolizzare i farmaci per poterli eliminare.
, che possono manifestarsi sia con l’antagonismo, e quindi una riduzione della risposta terapeutica, che con il sinergismo, con amplificazione della risposta terapeutica e, a volte ma non sempre, degli effetti collaterali. Come conseguenza di quanto detto, in sintesi, si può ragionevolmente affermare: (1) la politerapia non deve mai essere trattata con sufficienza, sopratutto se le combinazioni dei farmaci sono nuove; (2) a parità di efficacia clinica sarebbe preferibile utilizzare farmaci che presentano meno interazioni con altri farmaci. In effetti, è importante ricordare che una totale mancanza di interazione tra farmaci è rara. Infatti, l’interazione farmacocinetica tra farmaci può anche non essere diretta. Mettiamo il caso di una co-somministrazione di antibiotici e anticoagulanti orali. La flora batterica intestinale, in parte responsabile della produzione di vitamina K, risulta diminuita dal trattamento antibiotico, per cui l’anticoagulante, che per essere assorbito compete con la vitamina K, sarà assorbito in misura maggiore (oltre a poter anche esplicare una maggiore attività in assenza della vitamina). Il quadro globale, infine, è ulteriormente complicato dalla necessità dell’organismo di metabolizzare i farmaci per poterli eliminare..")
42
…con il verapamil, due farmaci che presentano interazioni con l’omeprazolo.

43
Tetracicline e cationi
e.g. Ca2+, Al3+, Mg2+ or Fe2+ . - Fe2+ Antacidi - Al3+, Mg2+ etc Latte, formaggio - Ca2+ Effetto significativo. e.g. gli antacidi possono ridurre l’assorbimento dell’80% Soluzione: Intervallo di somministrazione Esempi benefici: carbone attivo

44
Farmaci che alterano lo svuotamento gastrico
Analgesici (e.g. morfina petidina) Rallentamento sostanziale Antimuscarinici (e.g. atropina, propantelina) Rallentamento antidepressivi triciclici (e.g. imipramina) rallentamento Agonisti muscarinici (e.g. Bethanechol) Aumento motilita’
 Rallentamento sostanziale. Antimuscarinici (e.g. atropina, propantelina) Rallentamento. antidepressivi triciclici (e.g. imipramina) rallentamento. Agonisti muscarinici (e.g. Bethanechol) Aumento motilita’")
45
Significato clinico dose singola
Fast Un assorbimento ritardato porta ad un picco ematico minore e spostato nel tempo. Slow

46
Si ricorderà che la farmacocinetica si suddivide in 4 principali fasi: l’assorbimento, la distribuzione, il metabolismo e l’escrezione. Malgrado il principale livello di interazione tra farmaci nella clinica sia il metabolismo, è importante ricordare che un farmaco può modificare l’efficacia di un altro farmaco in tutte le fasi farmacocinetiche. Facciamo un esempio: se ad un paziente viene somministrato un farmaco che riduce la motilità intestinale, altre terapie concomitanti potranno ovviamente subirne delle conseguenze. Sempre a livello di assorbimento, farmaci che sono in grado di influenzare il pH del tratto gastrointestinale modificano l’assorbimento di un alto numero di farmaci modificandone sia il livello di ionizzazione che la solubilità. Un altro tipo di interazione importante può verificarsi a livello della distribuzione delle proteine plasmatiche. La maggior parte dei farmaci è attiva nella sua forma libera, cioè non legata alle proteine. Se un paziente assume due farmaci che si legano alla stessa proteina, la disponibilità di farmaco libero, soprattutto del farmaco che mostra una affinità minore per questa proteina, sarà ovviamente aumentata. L’azione di farmaci come il warfarin, ad esempio, che si legano molto intensamente alle proteine plasmatiche, è estremamente sensibile a questo tipo di interazione. Un altro esempio lampante in questo senso è costituito dal probenecid o l’alcalinizzazione delle urine. Abbiamo ovviamente citato casi limite, ma è ovvio che molti dei farmaci correntemente in uso interagiscono in una maniera o in un’altra con altri farmaci. Chiaramente, non sono state prese in considerazione le interazioni a livello di meccanismo d’azione (farmacodinamiche), che possono manifestarsi sia con l’antagonismo, e quindi una riduzione della risposta terapeutica, che con il sinergismo, con amplificazione della risposta terapeutica e, a volte ma non sempre, degli effetti collaterali. Come conseguenza di quanto detto, in sintesi, si può ragionevolmente affermare: (1) la politerapia non deve mai essere trattata con sufficienza, sopratutto se le combinazioni dei farmaci sono nuove; (2) a parità di efficacia clinica sarebbe preferibile utilizzare farmaci che presentano meno interazioni con altri farmaci. In effetti, è importante ricordare che una totale mancanza di interazione tra farmaci è rara. Infatti, l’interazione farmacocinetica tra farmaci può anche non essere diretta. Mettiamo il caso di una co-somministrazione di antibiotici e anticoagulanti orali. La flora batterica intestinale, in parte responsabile della produzione di vitamina K, risulta diminuita dal trattamento antibiotico, per cui l’anticoagulante, che per essere assorbito compete con la vitamina K, sarà assorbito in misura maggiore (oltre a poter anche esplicare una maggiore attività in assenza della vitamina). Il quadro globale, infine, è ulteriormente complicato dalla necessità dell’organismo di metabolizzare i farmaci per poterli eliminare.
, che possono manifestarsi sia con l’antagonismo, e quindi una riduzione della risposta terapeutica, che con il sinergismo, con amplificazione della risposta terapeutica e, a volte ma non sempre, degli effetti collaterali. Come conseguenza di quanto detto, in sintesi, si può ragionevolmente affermare: (1) la politerapia non deve mai essere trattata con sufficienza, sopratutto se le combinazioni dei farmaci sono nuove; (2) a parità di efficacia clinica sarebbe preferibile utilizzare farmaci che presentano meno interazioni con altri farmaci. In effetti, è importante ricordare che una totale mancanza di interazione tra farmaci è rara. Infatti, l’interazione farmacocinetica tra farmaci può anche non essere diretta. Mettiamo il caso di una co-somministrazione di antibiotici e anticoagulanti orali. La flora batterica intestinale, in parte responsabile della produzione di vitamina K, risulta diminuita dal trattamento antibiotico, per cui l’anticoagulante, che per essere assorbito compete con la vitamina K, sarà assorbito in misura maggiore (oltre a poter anche esplicare una maggiore attività in assenza della vitamina). Il quadro globale, infine, è ulteriormente complicato dalla necessità dell’organismo di metabolizzare i farmaci per poterli eliminare..")
48
Interazione tra farmaci
(e.g. albumina) 90% 80% Nuovo farmaco somministrato 10% 20% Warfarin Ibuprofen Fenitoina aspirina Furosemide e.g % legato 0.1% libero farmaco A 50.0% legato 50% libero farmaco B
 90% 80% Nuovo farmaco. somministrato. 10% 20% Warfarin. Ibuprofen. Fenitoina. aspirina. Furosemide. e.g. 99.9% legato 0.1% libero farmaco A. 50.0% legato 50% libero farmaco B.")
49
Fenitoina e cabamazepina
paz 1 2 3 tossico Range terapeutico Legame a proteine plasmatiche

50
Interazione albumina- farmaco

51
Interazione farmaco-albumina
(1 2 siti) Sito 1 Sito 2
 Sito 1. Sito 2.")
53
MICOTOSSINE La tossicità organospecifica (nel fegato) delle aminitina e falloidina deriva dalla penetrazione facilitata nell’epatocita delle tossine legate all’albumina. Quando le tossine sono legate all’albumina, la loro tossicità è aumentata di 10 volte circa. S.B.2003
 delle aminitina e falloidina deriva dalla penetrazione facilitata nell’epatocita delle tossine legate all’albumina. Quando le tossine sono legate all’albumina, la loro tossicità è aumentata di 10 volte circa. S.B")
54
TERAPIA DELLA INTOSSICAZIONE.2
Penicillina G (sodica) ad alte dosi 1 milione Unità/kg peso/die per via e.v. (ovviamente, fare prima il pomfo). Questo perché la penicillina si lega ad albumina, e compete per il legame con le tossine, spiazzando le tossine (N.B. : quando legate all’albumina, le micotossine sono 10 volte più potenti). Nei bambini, per labilità della BEE la penicillina può dare convulsioni; nei bambini si debbono usare dosi ridotte di penicillina. Nei pazienti allergici alla penicillina, si può usare la rifampicina, che però è meno efficiente in questo contesto. Terapia va protratta per 7-10 giorni S.B.2003
 ad alte dosi 1 milione Unità/kg peso/die per via e.v. (ovviamente, fare prima il pomfo). Questo perché la penicillina si lega ad albumina, e compete per il legame con le tossine, spiazzando le tossine (N.B. : quando legate all’albumina, le micotossine sono 10 volte più potenti). Nei bambini, per labilità della BEE la penicillina può dare convulsioni; nei bambini si debbono usare dosi ridotte di penicillina. Nei pazienti allergici alla penicillina, si può usare la rifampicina, che però è meno efficiente in questo contesto. Terapia va protratta per 7-10 giorni. S.B")
55
L’aspirina Fase I Fase II Idrolisi Metabolita glucuronidato
Ma anche a. salicilurico

56
Fase 1 Ossidazione Idrossilazione Dealchilazione Deaminazione Idrolisi Fase 2 Coniugazione con: glucuronato solfato metile acetato glicina

57
Ruolo degli enzimi CYP450 nel metabolismo dei farmaci
Enzima Farmaci metabolizzati (%) CYP2A6 3 CYP2B6 3 CYP2E1 4 CYP2C19 8 CYPA1/2 11 CYP2C8/9 16 CYP2D6 19 CYP3A4/5 36 Anche enzimi specifici: e.g. alcool deidrogenasi, colinesterasi Rogers JF et al. Am J Med 2002; 113:
 CYP2A6 3. CYP2B6 3. CYP2E1 4. CYP2C19 8. CYPA1/2 11. CYP2C8/9 16. CYP2D6 19. CYP3A4/5 36. Anche enzimi specifici: e.g. alcool deidrogenasi, colinesterasi. Rogers JF et al. Am J Med 2002; 113:")
58
Principali interazioni tra farmaci a livello del CYP
Inibizione veloce, spesso farmaco-specifica Induzione lenta, spesso non farmaco-specifica

59
Inibizione del CYP Competezione tra due substrati dello stesso CYP Competizione tra due sostanze di cui una non e’ metabolizzata dall’enzima Inattivazione dell’enzima Possibilita’ di potenziamento

60
Per riepilogare, quindi, se due farmaci si legano allo stesso enzima del citocromo P450, è possibile che uno dei due venga metabolizzato più lentamente. Se noi consideriamo una somministrazione ripetuta di un farmaco, tale situazione può evidentemente portare a delle conseguenze sicuramente rilevanti da un punto di vista clinico.

61
Induzione ed inibizione della P450
Induttori: etanolo, omeprazolo, fenobarbital, rifampicina, tabacco Inibitori: cimetidina, succo di pompelmo

62
CYP3A4

63
Enzyme Induction Time (hrs) No induction Phenobarbital induction
100 No induction Phenobarbital induction Zoxazolamine (mg/ml) Plasma Level (Classic barbiturate effect) 10 Benzopyrene induction (Generated from grilling meat) 1 Time (hrs)
 Plasma Level. (Classic barbiturate. effect) 10. Benzopyrene. induction. (Generated from. grilling meat) 1. Time (hrs)")
64
Meccanismo di induzione enzimatica
TCDD AhR Arnt CYP1A Barb. CAR RxR CYP2B Gene CYPx nucleo mRNA prot Tempo (giorni)
")
65
Possibili livelli di controllo dei livelli di CYP
mRNA pol 5’ AGGTCA AGGTCA NNN NNN TATA ATG 3’ 3’ 5’ stimoli tessutale xenobiotici basale Transcrizione Stabilita’ del mRNA Stabilita’ della proteina Proteina (CYP)
")
66
Induzione e regolazione di CYP3A
iperforina, ingrediente naturale dell‘erba di S. Giovanni (Johanniskraut, Hypericum perforatum) possiede elevata affinità per i PXR (Kd = 27 nM). Applicazioni: rimedio contro la colestasi, antidepressivo moderato. Interazioni: OCs (estroprogestinici): Figli dell‘iperico!!! Antivirali Viagra
 possiede elevata affinità per i PXR (Kd = 27 nM). Applicazioni: rimedio contro la colestasi, antidepressivo moderato. Interazioni: OCs (estroprogestinici): Figli dell‘iperico!!! Antivirali. Viagra.")
68
NECROSI EPATICA Glutathione Transferase Inactive

69
(Ahmad S. Diltiazem myopathy. Ann Heart J. 1993;126:1494-1495).
Case report (Ahmad S. Diltiazem myopathy. Ann Heart J. 1993;126: ). Un uomo di 53 anni con ipertensione, coronopatia e ipercolesterolemia era in trattamento con nitrati, enalapril e lovastatina. In seguito aggiunge diltiazem alla terapia. Avendo seguito il trattamento per alcuni mesi con questi farmaci, il paziente cominciò a manifestare segni e sintomi di miopatia. Egli lamentava dolore alle gambe ed alle braccia così violento da impedirgli di lavorare. Un elettromiogramma rivelò una miopatia con valori di CK di 4000 U/L. Una settimana dopo aver sospeso l'assunzione di lovastatina e diltiazem, la miopatia del paziente migliorò. Fu iniziata nuovamente la terapia con lovastatina senza alcun sintomo di miopatia. Quando fu reintrodotto in terapia il diltiazem i livelli di CK aumentarono nuovamente e comparve nuovamente la miopatia.
. Un uomo di 53 anni con ipertensione, coronopatia e ipercolesterolemia era in trattamento con nitrati, enalapril e lovastatina. In seguito aggiunge diltiazem alla terapia. Avendo seguito il trattamento per alcuni mesi con questi farmaci, il paziente cominciò a manifestare segni e sintomi di miopatia. Egli lamentava dolore alle gambe ed alle braccia così violento da impedirgli di lavorare. Un elettromiogramma rivelò una miopatia con valori di CK di 4000 U/L. Una settimana dopo aver sospeso l assunzione di lovastatina e diltiazem, la miopatia del paziente migliorò. Fu iniziata nuovamente la terapia con lovastatina senza alcun sintomo di miopatia. Quando fu reintrodotto in terapia il diltiazem i livelli di CK aumentarono nuovamente e comparve nuovamente la miopatia.")
70
(Ahmad S. Diltiazem myopathy. Ann Heart J. 1993;126:1494-1495).
Case report: commento (Ahmad S. Diltiazem myopathy. Ann Heart J. 1993;126: ). (basandosi sul razionale che la miopatia del paziente non si era presentata fino a quando il diltiazem non era stato aggiunto al regime terapeutico, l'autore attribuisce al diltiazem la responsabilità della miopatia) il diltiazem, inibendo l'isoforma 3A4 del citocroma P-450, provoca un innalzamento della concentrazione plasmatica della lovastatina l'attribuzione (errata) al diltiazem dell'evento indesiderato è un buon esempio della tendenza ad identificare l'ultimo farmaco aggiunto in terapia come quello nocivo la stessa attribuzione al diltiazem mette in evidenza quanto spesso siano sottovalutate le interazioni tra farmaci
. (basandosi sul razionale che la miopatia del paziente non si era presentata fino a quando il diltiazem non era stato aggiunto al regime terapeutico, l autore attribuisce al diltiazem la responsabilità della miopatia) il diltiazem, inibendo l isoforma 3A4 del citocroma P-450, provoca un innalzamento della concentrazione plasmatica della lovastatina. l attribuzione (errata) al diltiazem dell evento indesiderato è un buon esempio della tendenza ad identificare l ultimo farmaco aggiunto in terapia come quello nocivo. la stessa attribuzione al diltiazem mette in evidenza quanto spesso siano sottovalutate le interazioni tra farmaci.")
71
CYP1A2 Ad una donna di 63 anni in terapia di mantenimento con imipramina (100 mg/die) fu somministrato un altro antidepressivo, la fluvoxamina (100 mg/die), nel tentativo di potenziare la risposta. Dopo alcuni giorni comparvero tremore, secchezza delle fauci, costipazione e confusione, tipici effetti indesiderati da triciclici. L’interruzione del trattamento con fluvoxamina determinò la scomparsa di tali effetti. L’analisi delle co azioni plasmatiche di imipramina durante il trattamento con fluvoxamina evidenziarono che quest’ultima provocava un drammatico aumento delle concentrazioni plasmatiche di imipramina. Questo effetto era dovuto alla inibizione da parte della fluvoxamina della reazione di demetilazione della imipramina mediata dal CYP1A2, come dimostrato da studi in vitro. Spina E, Campo GM, Avenoso A, Pollicino MA and Caputi AP (1992) Interaction between fluvoxamine and imipramine/desipramine in four patients. Ther Drug Monit 14:
 Interaction between fluvoxamine and imipramine/desipramine in four patients. Ther Drug Monit 14:")
72
Metabolismo extraepatico

73
CYP3A4

74
Lovastatina come esempio

75
Succo di pompelmo Il succo di pompelmo, ma non quello d'arancia dolce, aumenta la biodisponibilità dei calcio-antagonisti. Nel caso della felodipina, che normalmente ha una biodisponibilità del 15% dopo metabolismo di primo passaggio, il succo di pompelmo produce concentrazioni plasmatiche di farmaco circa 3 volte più elevate della norma. Le conseguenze nei pazienti ipertesi borderline sono un'aumentata riduzione della pressione arteriosa ed un incremento della frequenza cardiaca. Le reazioni avverse correlate alla vasodilatazione (es. cefalea) sono di conseguenza più frequenti. Il succo di pompelmo inibisce selettivamente, nel tratto gastrointestinale, il CYP3A4. L'interazione tra felodipina e succo di pompelmo chiarisce due importanti concetti: l'importanza dell'intestino come sede di farmacometabolismo e che l'interazione dipende dalla via di somministrazione del farmaco. (Il succo di pompelmo non interagisce con farmaci somministrati per via endovenosa).
 sono di conseguenza più frequenti. Il succo di pompelmo inibisce selettivamente, nel tratto gastrointestinale, il CYP3A4. L interazione tra felodipina e succo di pompelmo chiarisce due importanti concetti: l importanza dell intestino come sede di farmacometabolismo e che l interazione dipende dalla via di somministrazione del farmaco. (Il succo di pompelmo non interagisce con farmaci somministrati per via endovenosa).")
76
Succo di pompelmo L’aritmia ventricolare (torsades de pointes) potenzialmente fatale da terfenadina, astemizolo e cisapride; il grave danno muscolo-scheletrico con insufficienza renale acuta (rabdomiolisi) da alcuni inibitori della HMG-CoA reduttasi, quali lovastatina, simvastatina, atorvastatina e cerivastatina; il danno renale da ciclosporina e tacrolimus; l'ipotensione sintomatica e le complicazioni ischemiche da calcio-antagonisti diidropiridinici, quali felodipina, nicardipina, nifedipina, nimodipina, nisolidipina, nitrendipina e pranidipina; l'eccessiva sedazione da benzodiazepine, quali midazolam e triazolam, e da buspirone; l'atassia da carbamazepina.
 da alcuni inibitori della HMG-CoA reduttasi, quali lovastatina, simvastatina, atorvastatina e cerivastatina; il danno renale da ciclosporina e tacrolimus; l ipotensione sintomatica e le complicazioni ischemiche da calcio-antagonisti diidropiridinici, quali felodipina, nicardipina, nifedipina, nimodipina, nisolidipina, nitrendipina e pranidipina; l eccessiva sedazione da benzodiazepine, quali midazolam e triazolam, e da buspirone; l atassia da carbamazepina.")
77
Williams et al, 2003

78
Williams et al, 2003

79
Farmacogenetica (e.g. polimorfismi del CYP450)
L’amitriptilina e’ un antidepressivo metabolizzato dal CYP2C19 Si e’ scoperto che geneticamente ciascuno di noi esprime uno dei seguenti CYP2C19 : A(lto) M(etabolismo), I(ntermediato) M and B(asso) M Quantita’ di amitriptilina in pazienti depressi: Tutti: AM: BM 3-4.1
 M(etabolismo), I(ntermediato) M and B(asso) M. Quantita’ di amitriptilina in pazienti depressi: Tutti: AM: BM")
80
Farmacocinetica tossicita’ [farmaco] effetti collaterali efficace
inefficace 1 2 3 4 5 6 7 8 9 10 tempo Assorbimento Distribuzione Metabolismo Eliminazione
![Farmacocinetica tossicita’ [farmaco] effetti collaterali efficace](http://slideplayer.it/slide/596600/2/images/80/Farmacocinetica+tossicita%E2%80%99+%5Bfarmaco%5D+effetti+collaterali+efficace.jpg "inefficace tempo. Assorbimento. Distribuzione. Metabolismo. Eliminazione.")
81
} Vie di escrezioni principali: Renale Epatica (bile) clearance
Polmonare Altro: latte materno, sudore, capelli, etc clearance Renale: Diffusione passiva (filtrazione glomerulare) Trasporto attivo Circolo entero-epatico
 Trasporto attivo. Circolo entero-epatico.")
82
EMIVITA DI UN FARMACO Il “tempo di dimezzamento”, “emivita” o t1/2 di un farmaco è il tempo necessario affinchè la concentrazione plasmatica di quel farmaco si riduca del 50% (ovvero, si dimezzi). E’ determinato dal volume di distribuzione (Vd) del farmaco e dalla sua clearance (Cl)
. E’ determinato dal volume di distribuzione (Vd) del farmaco e dalla sua clearance (Cl)")
83
Influenza di Vd e Cl sull’emivita di eliminazione di un farmaco
L’emivita è direttamente proporzionale al volume di distribuzione e inversamente proporzionale alla clearance. Una emivita più lunga è associata a una maggior volume di distribuzione e a una bassa clearance. T1/2 = 0.7 x Vd/Cl Esempio 1: Vd= 1000 l, Cl = 10 l/min. Allora T1/2 = 0.7 x (1000 l)/10 l/min = 70 minuti Esempio 2: Vd = 500 l, Cl = 25 l/min. Allora T1/2 = 0.7 x (500 l)/ 25 l/min = 14 minuti
/10 l/min = 70 minuti. Esempio 2: Vd = 500 l, Cl = 25 l/min. Allora. T1/2 = 0.7 x (500 l)/ 25 l/min = 14 minuti.")
84
EMIVITA DI UN FARMACO: 3 CONCETTI FONDAMENTALI
L’emivita serve per determinare quanto tempo sarà necessario per raggiungere lo “steady state” (stato stazionario; condizione di equilibrio) di un farmaco nel plasma. OCCORRONO 5-6 EMIVITE. Il tempo per raggiungere lo stato stazionario è indipendente dal dosaggio L’emivita serve a determinare quanto tempo occorrerà perché il farmaco sia totalmente eliminato dall’organismo dopo la somministrazione dell’ultima dose. Di nuovo, OCCORRONO 5-6 EMIVITE. L’emivita serve per determinare un corretto intervallo fra le somministrazioni.
 di un farmaco nel plasma. OCCORRONO 5-6 EMIVITE. Il tempo per raggiungere lo stato stazionario è indipendente dal dosaggio. L’emivita serve a determinare quanto tempo occorrerà perché il farmaco sia totalmente eliminato dall’organismo dopo la somministrazione dell’ultima dose. Di nuovo, OCCORRONO 5-6 EMIVITE. L’emivita serve per determinare un corretto intervallo fra le somministrazioni.")
85
PERCHE’ 5- 6 EMIVITE??? Prima emivita: 50% farmaco
Seconda emivita: 25% farmaco (la metà di 50) Terza emivita: 12.5% farmaco (la metà di 25) Quarta emivita: 6.2% farmaco (la metà di 12.5) Quinta emivita: 3.1 % farmaco (la metà di 6.2) Sesta emivita: 1.65% farmaco /la metà di 3.1) = quasi 99% farmaco
 Terza emivita: 12.5% farmaco (la metà di 25) Quarta emivita: 6.2% farmaco (la metà di 12.5) Quinta emivita: 3.1 % farmaco (la metà di 6.2) Sesta emivita: 1.65% farmaco /la metà di 3.1) = quasi 99% farmaco.")
86
Interazioni in farmacocinetica
Assorbimento pH gastrico ionizzazione Formazioni di complessi chelazione adsorbimento Alterazione nella motilita‘ gastrica Distribuzione Competizione per le stesse proteine plasmatiche Competizione per siti di legame nei tessuti bersaglio Metabolismo Author: Maton P & Burton M Title: Clinician’s manual on drug interactions in gastroenterology Source: Life Science Communication, London, UK Pharmacokinetic drug interactions can be divided on the basis of the site of interaction of the drugs. Interactions may influence absorption, distribution, metabolism or elimination of the drug. Escrezione modificazioni del circolo enteroepatico competizione per sistemi di trasporto alterazione del pH del tubulo distale Modificata da Maton & Burton; 1996

Presentazioni simili














:>")








