Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
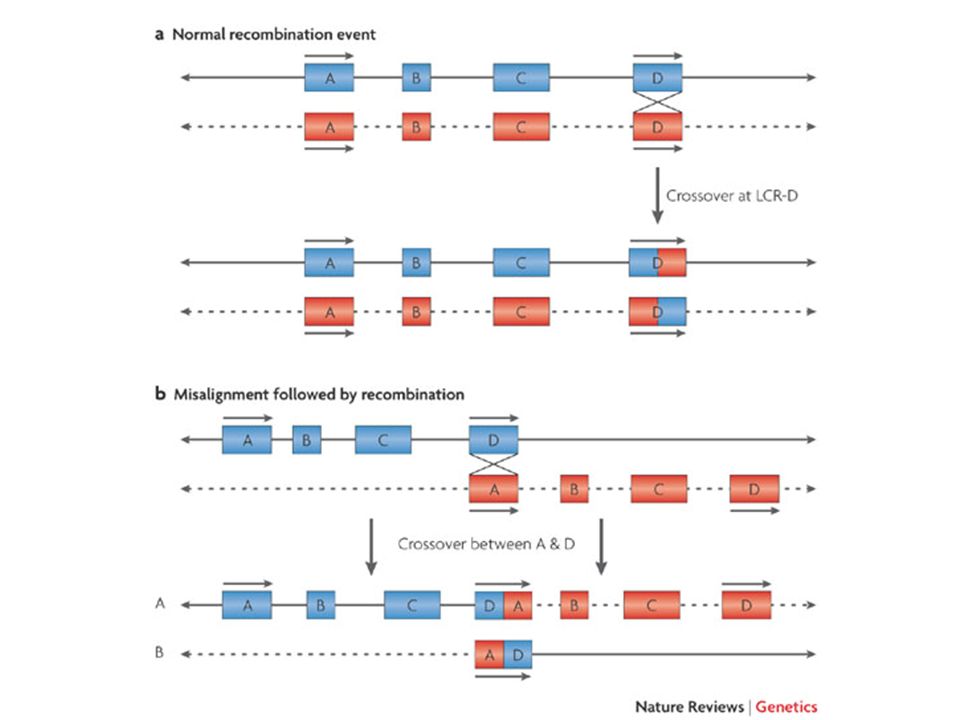
disordini genomici strutturali e submicroscopici
Vincenzo Nigro Dipartimento di Patologia Generale Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine (TIGEM)
")
2
portatori di un rischio riproduttivo indipendente dal partner
traslocazione reciproca portatori di una traslocazione cromosomica bilanciata (reciproca) scambio di materiale genetico tra cromosomi non omologhi non vi è modificazione della dose genica frequenza 1/520 nati fenotipicamente normale
 scambio di materiale genetico tra cromosomi non omologhi. non vi è modificazione della dose genica. frequenza 1/520 nati. fenotipicamente normale.")
3
traslocazioni bilanciate
lo scambio di segmenti cromosomici avviene senza perdita di alcuna informazione genetica nessuna regione cromosomica è assente, ma è solo trasferita su un altro cromosoma ma un gene di fusione tra due geni altrimenti separati, un evento che è comune nelle cellule maligne traslocazione reciproca

4
traslocazioni bilanciate (meiosi e fertilizzazione)
Traslocazione bilanciata Segregazione alternata Normale Traslocazione Segregazione adiacente 1 Traslocazione Trisomia Segregazione adiacente 2 Trisomia La formazione di tetravalenti aiuta a capire: solo con la segregazione alternata si formano gameti normali o con traslocazione bilanciata, mentre le segregazioni adiacenti 1 e 2 portano alla traslocazione sbilanciata o alla trisomia

5
traslocazioni robertsoniane (rob)
coinvolgono i cromosomi acrocentrici 13, 14, 15, 21 e 22 nessuna regione cromosomica è assente, perché questi contengono un braccio corto privo di geni che può risultare perduto con la fusione dei bracci q di due cromosomi acrocentrici La più frequente traslocazione Robertsoniana è la rob(13q14q) che rappresenta il 75% di tutte le rob segue poi la rob(14q21q) e la rob(21q21q) si formano in genere durante la meiosi femminile e comportano infertilità maschile o abortività ripetuta. rob
 che rappresenta il 75% di tutte le rob. segue poi la rob(14q21q) e la rob(21q21q) si formano in genere durante la meiosi femminile e comportano infertilità maschile o abortività ripetuta. rob.")
6
t(13;14) M=F 1% t(14;21) F 15% M 2% t(21;22) F 10% M 5%
Percentuale alla nascita di figli con cariotipo sbilanciato da genitori con traslocazione robertsoniana t(13;14) M=F 1% t(14;21) F 15% M 2% t(21;22) F 10% M 5% t(21;21) M=F 100%
 M=F 1% t(14;21) F 15% M 2% t(21;22) F 10% M 5% t(21;21) M=F 100%")
7
Traslocazione sbilanciata
maggiori sono le dimensioni cromosomiche, minore è la possibilità di una gravidanza a termine minori sono le dimensioni, maggiore è il rischio di un feto malformato Sesso del genitore donna>uomo (gli spermatozoi hanno il 7.5% di difetti contro l’1% degli oociti, ma sono selezionati) Il rischio aumenta se il difetto è stato accertato a partire da un figlio precedente con cariotipo sbilanciato
 Il rischio aumenta se il difetto è stato accertato a partire da un figlio precedente con cariotipo sbilanciato.")
8
rischio alla nascita di figli con cariotipo sbilanciato
Se non vi sono stati casi in famiglia e la madre è eterozigote per una traslocazione reciproca il rischio è il 7% Se non vi sono stati casi in famiglia e il padre è eterozigote per una traslocazione reciproca il rischio è il 3% Se vi sono stati casi di traslocazioni sbilanciate in famiglia e la madre è eterozigote il rischio è il 14% Se vi sono stati casi di traslocazioni sbilanciate in famiglia e il padre è eterozigote il rischio è l’8%

9
inversioni Le inversioni sono rare (meno di 1 caso su 1000) e a volte difficili da mettere in evidenza Possono essere semplici quando comprendono due punti di rottura su di un singolo cromosoma Sono pericentriche quando il segmento invertito contiene il centromero (es: 46, XX inv(3)p25q21) Le inversioni pericentriche dei cromosomi 1, 9, 16 e Y sono eteromorfismi citogenetici di normale riscontro in soggetti sani Le inversioni sono dette paracentriche se confinate ad uno dei due bracci (es: 46,XX. Inv(11)q21q23) L’eterozigote per un’inversione è un soggetto normale.
p25q21) Le inversioni pericentriche dei cromosomi 1, 9, 16 e Y sono eteromorfismi citogenetici di normale riscontro in soggetti sani. Le inversioni sono dette paracentriche se confinate ad uno dei due bracci (es: 46,XX. Inv(11)q21q23) L’eterozigote per un’inversione è un soggetto normale.")
10
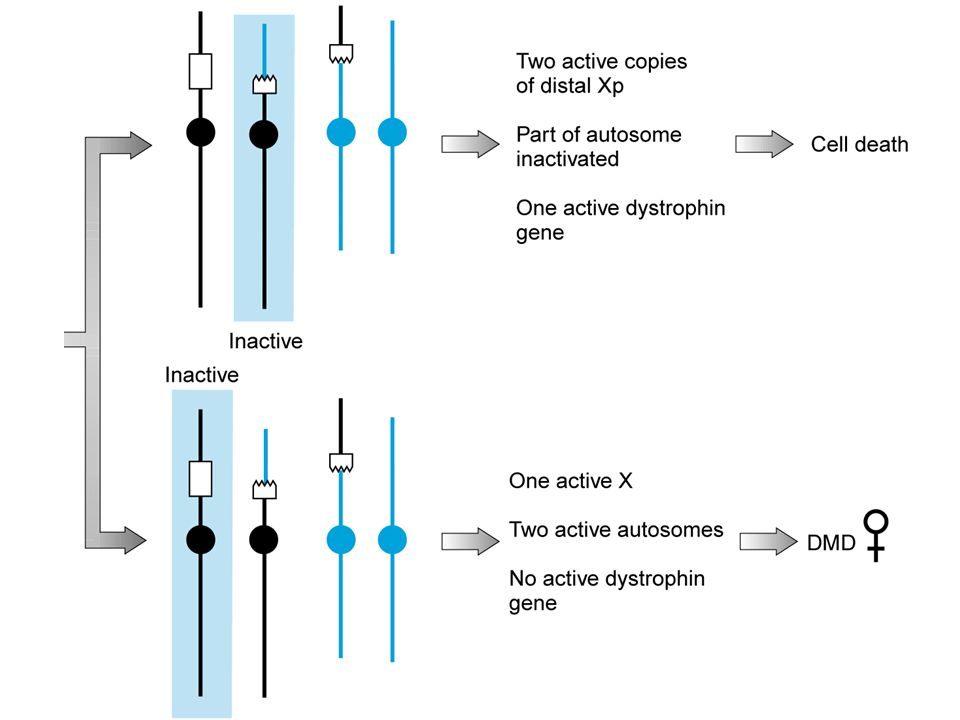
maschi sterili, femmine inattivano la X normale
coppia con familiarità per anomalie cromosomiche è indicazione all’esecuzione di un cariotipo fetale e l’estensione dell’indagine ai parenti traslocazioni X-autosoma maschi sterili, femmine inattivano la X normale traslocazioni robertsoniane non 21 60% cariotipo bilanciato 21 15% rischio di Down, se è eterozigote la madre 1% se è eterozigote il padre inversioni pericentriche varianti dell’1, 9, 16 e Y, in altri casi il rischio è 5-10% paracentriche, rischi inferiore allo 0.5%

11
donna eterozigote per una traslocazione bilanciata X-autosoma

13
disordine genomico submicroscopico
un disordine genomico submicroscopico è una patologia causata da acquisizione perdita alterazione di uno o più geni contigui le cui variazioni di dosaggio possono produrre effetti fenotipici La base molecolare è rappresentata da riarrangiamenti genomici, quali duplicazioni, delezioni, inversioni, senza alterazioni apparenti del cariotipo (<5Mb)
")
14
dominanza e recessività
in genetica, il carattere (o l’allele) è dominante se l’eterozigote è indistinguibile dall’omozigote in medicina la malattia è: dominante: fenotipo clinicamente manifesto con 1 allele mutato recessiva: fenotipo clinicamente manifesto con 2 alleli mutati (omozigote o eterozigote composto)
 è dominante se l’eterozigote è indistinguibile dall’omozigote. in medicina la malattia è: dominante: fenotipo clinicamente manifesto con 1 allele mutato. recessiva: fenotipo clinicamente manifesto con 2 alleli mutati (omozigote o eterozigote composto)")
15
La maggior parte dei geni autosomici si trova nella condizione A o C: il dosaggio genico critico è <50%. In tal caso, si osserva un fenotipo patologico solo se entrambi gli alleli sono colpiti I geni autosomici responsabili della patogenesi dei disordini genomici si trovano nella condizione B o D: si osserva un fenotipo già in eterozigosi per aploinsufficienza. Spesso anche un dosaggio genico aumentato >>100% può determinare una patologia

16
aploinsufficienza insufficiente quantità di prodotto genico causata da una mutazione in eterozigosi la mutazione è di tipo allele amorfo o ipomorfo colpisce geni per i quali il 50% di prodotto genico non è abbastanza per garantirne la funzione spesso un dosaggio preciso è richiesto ai fattori di trascrizione e alle molecole di segnale espressi nel corso dello sviluppo

17
In caso di delezioni del cromosoma X nei maschi si osserva direttamente in fenotipo come sindrome da geni contigui In caso di delezioni autosomiche in eterozigosi, molto spesso il dosaggio dimezzato non è causa di malattia. Quando si osserva una sindrome da delezione, è risolutivo trovare la stessa sindrome causata da una mutazione puntiforme in uno solo dei geni. Se questa non si trova, la sindrome esiste solo come somma di più difetti.

18
ACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGAACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGA

19
10% of the human genome could vary in copy number
ACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGAACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACTATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGA Copy Number Variation 10% of the human genome could vary in copy number 1 2

20
duplicazioni segmentali
il genoma umano contiene complessivamente il 13,7% di segmenti duplicati con almeno il 90% di identità di sequenza il 5,2% del genoma contiene segmenti duplicati lunghi tra 1 e 10kb, mentre il 4,5% tra 10kb e 20kb i cromosomi più colpiti sono l’Y (50,4%) ed il 22 (11,9%), ma anche il 7, 9, 10, 15, 16, 17 e X le duplicazioni segmentali possono essere intracromosomiche o intercromosomiche con tre localizzazioni differenti: pericentromeriche (47Mb, dupliconi originati da altri cromosomi) subtelomeriche (ciascuna solo kb, orientate) interstiziali (solo nella specie umana sono disseminate ad una distanza media di 3Mb)
 ed il 22 (11,9%), ma anche il 7, 9, 10, 15, 16, 17 e X. le duplicazioni segmentali possono essere intracromosomiche o intercromosomiche. con tre localizzazioni differenti: pericentromeriche (47Mb, dupliconi originati da altri cromosomi) subtelomeriche (ciascuna solo kb, orientate) interstiziali (solo nella specie umana sono disseminate ad una distanza media di 3Mb)")
22
Malattie autosomiche dominanti
Come fanno le delezioni in uno solo dei due alleli a costituire un carattere dominante? il livello dimezzato di prodotto genico è insufficiente a mantenere il fenotipo il difetto eterozigote diviene omozigote a livello delle cellule dei tessuti periferici (LOH) un solo allele è espresso per imprinting dell’altro
 un solo allele è espresso per imprinting dell’altro.")
23
FISH

24
Sonde FISH le sonde FISH devono essere mirate, non possono essere utili nell’analisi genomica generale telomero centromero intero cromosoma locus

25
FISH

26
Sindrome di DiGeorge

27
DiGeorge/velocardiofacciale
La sindrome di DiGeorge del22q11.2 è la più frequente sindrome da microdelezione, con un incidenza di 1 su 4000—5000 nati La delezione comprende 3Mb ed almeno 30 geni

28
La migrazione di cellule della cresta neurale contribuisce alle strutture embrionali colpite nella sindrome di DiGeorge

29
DiGeorge È caratterizzata da Anomalie cardiache T-cell deficit
palatoschisi anomalie facciali Ipocalcemia Mutazioni puntiformi del gene TBX1 possono portare a questi 5 tratti fenotipici, ma non alle difficoltà nell’apprendimento che è invece frequente nella sindrome da delezione

30
Williams-Beuren prevalenza alla nascita 1/7500-1/20.000, ma può non essere diagnosticata

31
Williams una delezione tipica

32
Williams genetica gene dell’elastina LIM kinase 1 (LIMK1)
delezione “de novo” trasmissione autosomica dominante delezione di 1.6MB da 21 geni contigui in eterozigosi a 7q11.23 gene dell’elastina LIM kinase 1 (LIMK1) CLIP-115 che lega i microtubuli Fattori di trascrizione GTF2I e GTF2IRD1 effetto posizionale su altri geni circostanti la delezione
 CLIP-115 che lega i microtubuli. Fattori di trascrizione GTF2I e GTF2IRD1. effetto posizionale su altri geni circostanti la delezione.")
33
FISH della s. di Williams delezione 7q11.23
rilevabile mediante FISH ma non cariotipo

34
Williams comportamento
lieve o medio ritardo mentale (IQ tra 41 e 80) scarsa capacità di concentrazione ritardo nell’apprendimento del linguaggio e poi esagerata loquacità personalità amichevole e affettuosa danno facilmente confidenza anche a sconosciuti ansietà, spesso preoccupati per il benessere altrui ipersensibilità ai suoni memoria visiva e uditiva spesso fuori dal comune ricordano persone, luoghi e motivi musicali predisposizione ad imparare le lingue e la musica
 scarsa capacità di concentrazione. ritardo nell’apprendimento del linguaggio e poi esagerata loquacità. personalità amichevole e affettuosa. danno facilmente confidenza anche a sconosciuti. ansietà, spesso preoccupati per il benessere altrui. ipersensibilità ai suoni. memoria visiva e uditiva spesso fuori dal comune. ricordano persone, luoghi e motivi musicali. predisposizione ad imparare le lingue e la musica.")
35
Williams aspetto e segni
Faccia da elfo Occhi blu (77%) con pattern stellato dell’iride (74%) ma questo vale per i nordeuropei, strabismo (40%) Naso con la punta bulbosa bocca larga e guance piene microdontia e micrognazia Statura 10 cm in meno del normale ipercalcemia stenosi periferica delle arterie polmonari stenosi aortica sopravalvolare
 con pattern stellato dell’iride (74%) ma questo vale per i nordeuropei, strabismo (40%) Naso con la punta bulbosa. bocca larga e guance piene. microdontia e micrognazia. Statura 10 cm in meno del normale. ipercalcemia. stenosi periferica delle arterie polmonari. stenosi aortica sopravalvolare.")
36
Williams foto

37
Williams foto

38
Wolf-Hirschhorn genetica
delezione “de novo” di circa 4MB le delezioni sono più frequenti nella linea germinale maschile trasmissione autosomica dominante Regione critica di 165 kb di molti geni contigui in eterozigosi a 4p16.3

39
Wolf-Hirschhorn delezione a 4p16.3

40
Wolf-Hirschhorn Scarso accrescimento Ritardo mentale, ipotonia
Labbro leporino Conformazione ad elmo di guerriero greco

41
Sindrome 5p- (cri du chat) 1:50.000 nati
Pianto acuto e flebile Caratteristiche principali: Ritardo di crescita Microcefalia ed ipertelorismo Ipotonia, diastasi dei retti Deficit intellettivo e del linguaggio

42
imprinting una piccola parte del genoma umano presenta differenze significative nell’espressione a seconda se un gene è ereditato dal padre o dalla madre se due cromosomi sono provenienti dallo stesso genitore (disomia uniparentale) e non da entrambi i genitori, si determina morte o malattia alla base di queste differenze c’è l’imprinting genomico è dovuto a modificazioni epigenetiche (non correlate alla sequenza primaria del DNA) comporta l'aggiunta di un gruppo metile (CH3)- alla posizione 5 di un residuo di citosina nel DNA che precede immediatamente un residuo di guanina (CpG).
 e non da entrambi i genitori, si determina morte o malattia. alla base di queste differenze c’è l’imprinting genomico. è dovuto a modificazioni epigenetiche (non correlate alla sequenza primaria del DNA) comporta l aggiunta di un gruppo metile (CH3)- alla posizione 5 di un residuo di citosina nel DNA che precede immediatamente un residuo di guanina (CpG).")
43
Imprinting Figure 1. Imprint establishment and propagation during gametogenesis and development. The paternal allele (dashed line) is imprinted and the maternal allele is expressed (solid line). The "imprint mark" (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both "imprint marks" and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate "imprint marks" are reestablished for the next generation Am J Pathol 1999 Mar;154(3):635-47 Genomic Imprinting: Implications for Human Disease J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*
 is imprinted and the maternal allele is expressed (solid line). The imprint mark (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both imprint marks and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate imprint marks are reestablished for the next generation. Am J Pathol 1999 Mar;154(3): Genomic Imprinting: Implications for Human Disease. J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*")
44
Imprinting Nelle cellule germinali primordiali l’imprinting viene cancellato del tutto e il DNA è demetilato Successivamente nella linea germinale maschile si determina un pattern di imprinting che in alcuni loci è complementare a quello della linea germinale femminile I cromosomi su cui avviene l’imprinting (7, 11, 15) manterranno questo pattern e lo riprodurranno ad ogni mitosi Si potranno sempre distinguere l’espressione genica del cromosoma materno e paterno Figure 1. Imprint establishment and propagation during gametogenesis and development. The paternal allele (dashed line) is imprinted and the maternal allele is expressed (solid line). The "imprint mark" (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both "imprint marks" and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate "imprint marks" are reestablished for the next generation Am J Pathol 1999 Mar;154(3):635-47 Genomic Imprinting: Implications for Human Disease J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*
 manterranno questo pattern e lo riprodurranno ad ogni mitosi. Si potranno sempre distinguere l’espressione genica del cromosoma materno e paterno. Figure 1. Imprint establishment and propagation during gametogenesis and development. The paternal allele (dashed line) is imprinted and the maternal allele is expressed (solid line). The imprint mark (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both imprint marks and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate imprint marks are reestablished for the next generation. Am J Pathol 1999 Mar;154(3): Genomic Imprinting: Implications for Human Disease. J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*")
45
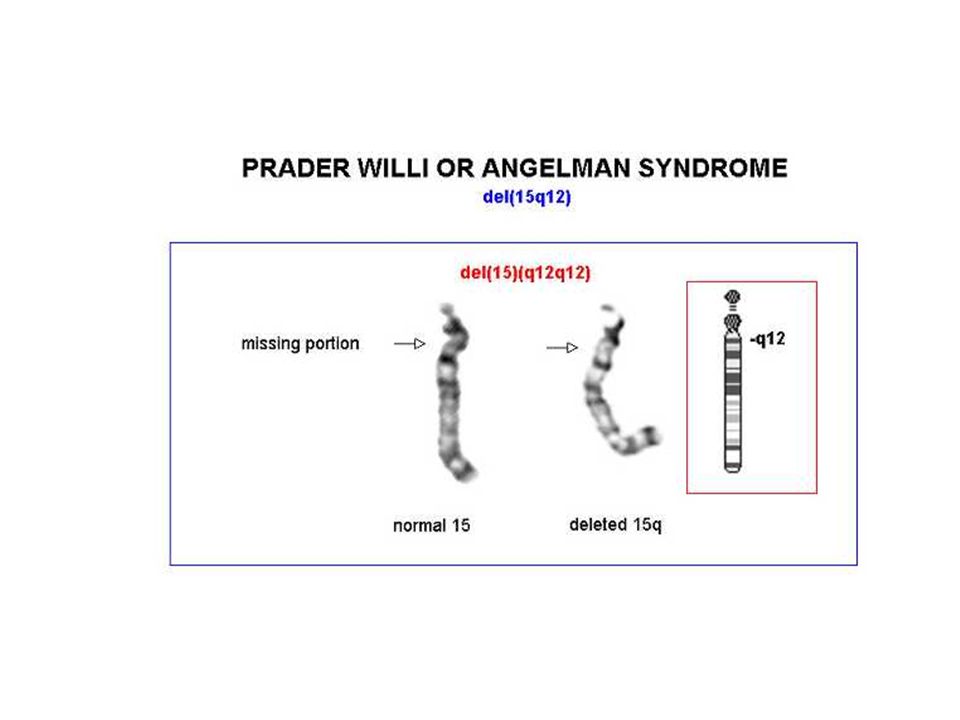
Disomia uniparentale Due copie dello stesso cromosoma sono ereditate dallo stesso genitore Spesso questo avviene attraverso un fenomeno transitorio di trisomia, seguito dalla perdita del cromosoma singolo e mantenimento del cromosoma doppio

46
Angelman delezione disomia uniparentale paterna
70% dei casi delezione della regione cromosomica 15q11-q13, che è soggetta al fenomeno dell'imprinting del cromosoma paterno Il gene materno (l'unico espresso) può essere alterato con 4 meccanismi noti: delezione disomia uniparentale paterna difetti nell'imprinting mutazioni a carico del gene UBE3A (ubiquitin ligasi) La diagnosi è clinica e il difetto genetico non si identifica nel 20% dei casi
 può essere alterato con 4 meccanismi noti: delezione. disomia uniparentale paterna. difetti nell imprinting. mutazioni a carico del gene UBE3A (ubiquitin ligasi) La diagnosi è clinica e il difetto genetico non si identifica nel 20% dei casi.")
48
Angelman "happy puppet syndrome" si può identificare in Cucciolo (Dopey) "addormentato", il più giovane dei nani che non ha mai imparato a parlare ritardo mentale con assenza del linguaggio, difficoltà nell'equilibrio, eccessivo buon umore

49
Angelman L'incidenza è 1/20.000 nati
crisi epilettiche e comunque alterazioni dell'EEG e microcefalia relativa

50
Prader-Willi iperfagia>obesità eccessiva assunzione di liquidi reazioni abnormi ai sedativi acromicria, criptorchidismo insensibilità al dolore, lesioni cutanee sbalzi di umore

51
Prader-Willi 1/15.000

52
Nomenclatura delle delezioni
Le delezioni sono designate con la sigla del che segue i numeri dei nucleotidi a monte e a valle della delezione separatida un segno _ 82_83del (o 82_83delTG) indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC
 indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC.")
53
Cosa sono le distrofie muscolari?
Malattie degenerative progressive Variazione dello spessore delle miofibrille con forti cambiamenti nella istologia del muscolo indebolimento e degenerazione del tessuto muscolare in fibroso e adiposo aree di necrosi con processi infiammatori

54
Duchenne Becker cingoli Emery- Dreifuss distale facio-scapolo- omerale oculo- faringea

55
Distrofia muscolare Duchenne/Becker
DMD Duchenne - 1/3,500 maschi Insorgenza -- Infanzia - tra 2 e 6 anni Sintomi – Debolezza generalizzata e danno muscolare prima agli arti e al tronco, polpacci ingrossati Progressione – Lenta ma inesorabile. Colpisce tutti i muscoli volontari. Sopravvivenza fino a anni BMD Becker - 1/10,000 maschi Insorgenza – Adolescenza o dopo Sintomi – Identici alla DMD ma più attenuati. Vi è coinvolgimento cardiaco significativo Progressione – Più lenta e più variabile della distrofia di Duchenne con buona aspettativa di vita

56
Le delezioni intrageniche del gene della distrofina mandano fuori cornice la lettura delle triplette quando gli esoni cancellati contenevano un numero di nucleotidi che non è multiplo esatto di tre (1,2,4,5,7,8,10,11 ecc). Questo causa la distrofia di Duchenne.

58
Le delezioni intrageniche che non alterano la cornice di lettura portano alla distrofia muscolare di Becker o ad un apparente buona salute. Forniscono informazioni per preparare delle microdistrofine per la terapia genica

59
Nomenclatura delle delezioni
Le delezioni sono designate con la sigla del che segue i numeri dei nucleotidi a monte e a valle della delezione separatida un segno _ 82_83del (o 82_83delTG) indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC
 indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC.")
60
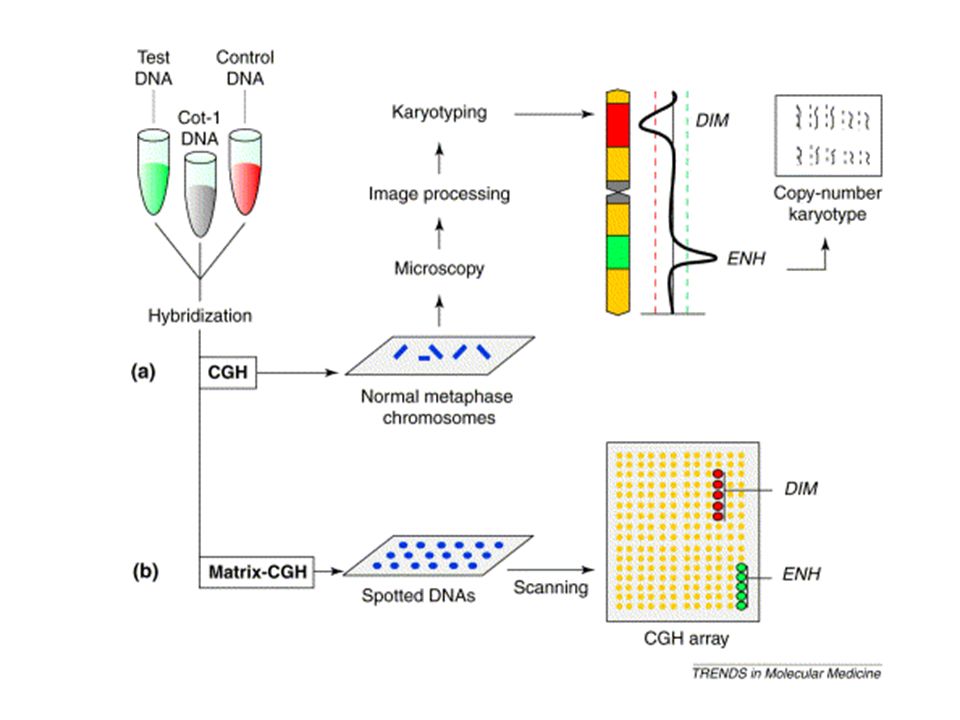
La tecnica del CGH (comparative genomic hybridization) permette l’individuazione di sequenze delete o duplicate nel genoma da testare (red) mediante il confronto con un genoma di riferimento (green). Sono preparate due sonde fluorescenti di colore diverso che ibridano contemporaneamente sui cromosomi. Se in una regione cromosomica prevale il colore (green) relativo al genoma di controllo questo significa che il genoma da testare (red) ha una delezione in quella regione
 relativo al genoma di controllo questo significa che il genoma da testare (red) ha una delezione in quella regione.")
62
Analisi delle immagini con software dedicati
DNA del paziente DNA di controllo Aggiungere DNA Cot-1 Ibridazione CGH CGH array Analisi delle immagini con software dedicati

63
Transcription Factors
Agilent Technology DNA RNA From one-dimensional to multi-dimensional … with multiple applications … Splice Variants ChIP GX aCGH/ CNV CH3 miRNA Sure Select Elucidate the role that protein-DNA interactions play in transcription, replication, modification and repair Perform global interrogations of the transcriptome and identify alternative splice forms to uncover the role gene variants play in drug response and disease Explore gene transcription on a genome-wide basis across a variety of model systems mRNA Conduct high-resolution, genome-wide profiling of DNA copy number changes associated with cancer and other genetic diseases Copy number Discover and monitor epigenetic modifications known to play a fundamental role in many cellular processes Methylation Transcription Factors mRNA isoforms Profile microRNAs and explore the role they play in gene regulation RNA interference DNA Target Enrichment Gene Silencing Protein Mutagenesis Whole Gene Synthesis Genome Partitioning Piattaforma selezionata e ventaglio applicativo.

64
Evoluzione del catalogo delle sonde già testate “Human CGH High Density (HD) Database”
combine with new database (6M probes) V1 2005 combine with two new databases (18M + 4M) V2 2006 V3 2008 4 million, 400bp spacing Scoring model v1 Probes pass Agilent Tm and homology filters One mapping per probe 8 million, 200bp spacing Scoring model v2 Used in selection of 244K and 105K CGH catalog products 24 million, 40bp spacing Scoring model v3 Probes pass Agilent Tm filters One or more mappings per probe Used for selection of new SurePrint G3 catalog designs Probe with >40 perfect matches marked as unmapped La crescita del database Agilent fino all’attuale versione.
 V combine with two new databases (18M + 4M) V V million, 400bp spacing. Scoring model v1. Probes pass Agilent Tm and homology filters. One mapping per probe. 8 million, 200bp spacing. Scoring model v2. Used in selection of 244K and 105K CGH catalog products. 24 million, 40bp spacing. Scoring model v3. Probes pass Agilent Tm filters. One or more mappings per probe. Used for selection of new SurePrint G3 catalog designs. Probe with >40 perfect matches marked as unmapped. La crescita del database Agilent fino all’attuale versione.")
65
Agilent Manufacturing Facility
Industrial manufacturing – Class 10,000 clean-room Ability to easily scale production capacity FAB wired directly into “e-array” allowing direct customer access to fully customizable microarrays High-performance Ink Jet Printing Enables High Levels of Spatial Multiplexing and Flexible Designs Advances to printing technology continue to occur against which Agilent can capitalize upon without having to make significant investments. How to Use Microarrays to Enhance Your Cancer Research 65

66
Formati dei Microarray Agilent
New SurePrint G3 Arrays 1M 400K 180K 60K nuovi 22K 44K 1.9K 11K Multiplex vecchi 244K 44K 105K 15K attuali 2005 2006 2007 2008 2001 2002 2003 2004 Evoluzione dei microarray agilent e formato che intendiamo utilizzare.

67
digestion with AluI and Rsa I and labeling
44,000-oligonucleotide with 8,769 interrogating probes (60-mer oligonucleotides) Agilent Technologies average spacing of probes across the DMD coding region of at least one every 144 base pairs digestion with AluI and Rsa I and labeling Agilent CGH-Analytics V3.4s software F F
 Agilent Technologies. average spacing of probes across the DMD coding region of at least one every 144 base pairs. digestion with AluI and Rsa I and labeling. Agilent CGH-Analytics V3.4s software. F. F.")
68
9293 probes that were replicated twice (Agilent)
remaining spots on each 44 K array were filled with probes from the X chromosome (11745) and all of the autosomes (12053) CGH Analytics software (v3.5)
 and all of the autosomes (12053) CGH Analytics software (v3.5)")
69
microarray-based mutation detection in the dystrophin gene (Nimblegen)
385,474 probes spanning the 2,222,000 bases of the dystrophin gene on chromosome X: 31,046,000–33,268,000 probe lengths from 45 to 60 nt with isothermal melting temperature DNA samples sonicated to a size between 500 and 2,000 bases Labelling with Klenow and Cy3 or Cy5 Exon 44

70
(SegMNTor DNA copy) NimbleScan
Normalized log2 ratio data were analyzed using two different analysis programs: (SegMNTor DNA copy) NimbleScan Gain and Loss Analysis of DNA (GLAD) ( DNA copy dup 2-4 GLAD DNA copy del 44 GLAD
 NimbleScan. Gain and Loss Analysis of DNA (GLAD) ( DNA copy. dup 2-4. GLAD. DNA copy. del 44. GLAD.")
71
(1, gain; 0, normal; and–1, loss),
del 17-44 del 48-52

72
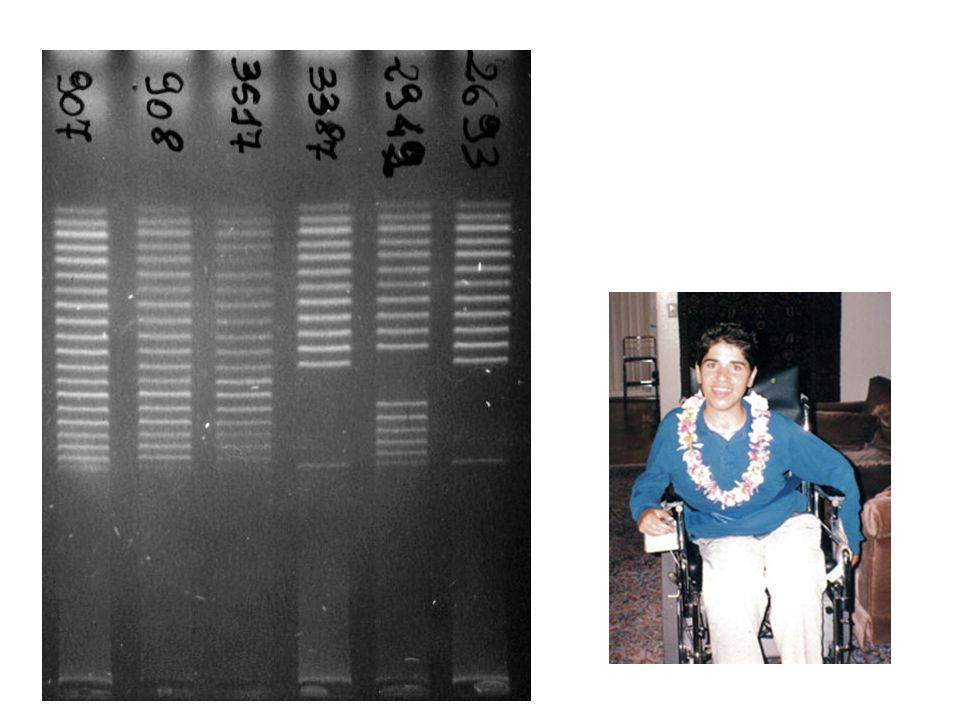
Solo la CGHarray riesce a chiarire la mutazione nelle donne portatrici di una delezione o duplicazione del cromosoma X. A causa della presenza del secondo cromosoma X normale sono difficili da identificare con altre tecniche del 46-55 del 49-50 dup 18-38

Presentazioni simili













 DI BREVI TRATTI RIPETUTI>")





