Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
ESEMPIO DI DETERMINAZIONE DELLA STRUTTURA DI UN POLISACCARIDE
Il caso dell’ esopolisaccaride prodotto da Enterobacter amnigenus: un batterio isolato dalle barbabietole da zucchero Ref. Paola Cescutti, Anne Kallioinen, Giuseppe Impallomeni, Renato Toffanin, Piero Pollesello, Matti Leisola e Tero Eerikäinene. “Structure of the exopolysaccharide produced by Enterobacter amnigenus”. Carbohydrate Research (2005) 340:439–447
 340:439–447.")
2
Presenza di polisaccaridi sulla superficie batterica
(caso dei Gram-negativi)
")
3
La prima cosa da fare è coltivare il batterio su un opportuno terreno di coltura: in questo caso estratto di lievito con frammenti peptidici e sali opportuni.

4
Man mano che le colonie batteriche crescono e producono il polisaccaride la viscosità del mezzo cresce. Misurando la viscosità si ha un’idea della produzione dell’esopolisaccaride. La purificazione del prodotto si esegue mediante centrifugazione, precipitazione con 4 volumi di etanolo, ri-dissoluzione in acqua e dialisi contro acqua.

5
Schema del funzionamento di una dialisi per separare grandi molecole dalle piccole molecole

6
E’ necessario controllare se la purificazione ha eliminato anche la maggior parte di proteine e DNA provenienti da cellule batteriche lisate. Si può utilizzare la spettroscopia UV.

7
A volte è necessario utilizzare tecniche più sensibili come il dicroismo circolare che permette di “vedere” solo molecole chirali.

8
Si procede con un’idrolisi del polimero in TFA 2M a 125°C per 1 h.
A questo punto si può procedere con l’analisi di composizione in zuccheri neutri. Si procede con un’idrolisi del polimero in TFA 2M a 125°C per 1 h. I monosaccaridi ottenuti sono ridotti ad alditoli con boroidruro di sodio effettuando poi un’acetilazione dei prodotti. Attenzione: la riduzione del fruttosio produce mannitolo e glucitolo

9
L’analisi dei prodotti acetilati è effettuata con GC su colonna SP2330 (a polarità intermedia) con un programma di temperatura da 200° a 245°C a 4°/min con elio come gas di trasporto ed utilizzando un rivelatore ad ionizzazione di fiamma.
 con un programma di temperatura da 200° a 245°C a 4°/min con elio come gas di trasporto ed utilizzando un rivelatore ad ionizzazione di fiamma.")
10
Qui salteremo per ora alle conclusioni
Per capire la tabella precedente si deve aver presente la struttura del polimero in esame. Qui salteremo per ora alle conclusioni [3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n 3 1 -D-Galp -D-GlcAp 4 -D-Manp CH COOH Il fucosio è piuttosto labile così come il mannosio legato ad un acido uronico.
--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n. 3. 1. -D-Galp. -D-GlcAp. 4. -D-Manp CH3 COOH. Il fucosio è piuttosto labile così come il mannosio legato ad un acido uronico.")
11
La seconda parte dell’analisi di composizione riguarda la determinazione della presenza di acidi uronici. Si effettua attraverso una metanolisi con HCl 1 M in MeOH a 85°C per 18 h. Si preparano poi i trimetil-silil derivati (con esametildisilazano + trimetilclorosilano +piridina per 1h a T amb). Si esegue quindi una gas-cromatografia su colonna HP1 (idrofobica, 50 m) a 150°C per 1 min e 150°C – 280°C a 3°C/min, infine 280°C per 20 min. Ogni saccaride dà più di un segnale perché non si riduce l’aldeide e quindi si ha un equilibrio tra tutti gli isomeri, ciclici e no.
 a 150°C per 1 min e 150°C – 280°C a 3°C/min, infine 280°C per 20 min. Ogni saccaride dà più di un segnale perché non si riduce l’aldeide e quindi si ha un equilibrio tra tutti gli isomeri, ciclici e no.")
12
Oltre a confermare la presenza dei saccaridi già trovati, viene determinata la presenza di acido glucuronico Fuc Gal Man Glc GlcA

13
E’ necessario verificare se l’enantiomeria dei saccaridi nel polimero è L o D: Si effettua una metanolisi + butanolisi con un alcol chirale: (+)-2-butanolo. Poi si procede come per l’analisi degli uronici: trimetilsililazione e GC su HP1. Il paragone con composti standard permette di risolvere il problema. Nel nostro caso sono tutti D meno il fucosio che è L (che è il caso più comune come anche per il ramnosio). L-Fuc D-Man D-Gal D-Glc D-GlcA
. L-Fuc. D-Man. D-Gal. D-Glc. D-GlcA.")
14
A questo punto si passa alla determinazione dei legami glicosidici per sapere i vari monosaccaridi come sono connessi tra loro (alfa, beta, 1-2, 1-3, 1-4, 1-6)
")
15
Si favorisce la dissociazione degli OH alcolici utilizzando idruro di potassio in DMSO (dimsile di potassio). Si permetilano le funzioni alcolato con CH3I. Si estraggono i saccaridi permetilati con acqua/cloroformio (si ritiene la frazione organica). Poi si effettua il procedimento già utilizzato per gli aditoli acetilati. Si ottengono così gli alditoli metilati (sulle posizioni degli OH liberi) e acetilati (sulle posizioni impegnate in legami glicosidici e sull’ossidrile in 5 impegnato nell’emiacetale.
 e acetilati (sulle posizioni impegnate in legami glicosidici e sull’ossidrile in 5 impegnato nell’emiacetale.")
16
Polisaccaride Polisaccaride metilato sugli OH liberi Idrolisi, riduzione ed acetilazione degli OH liberi

17
Si utilizza la spettrometria di massa ad impatto elettronico per analizzare i segnali risultanti dall’analisi GC. Le frammentazioni sono diagnostiche per individuare dove c’è metilazione e dove c’è acetilazione in quanto la frammentazione è più facile sui legami CC tra due gruppi metossile o metossile/acetile mentre non avviene tra due C sostituiti con acetile. Inoltre la carica positiva rimane sul frammento che porta il gruppo metile.

18
a) I numeri indicano la posizione dei legami glicosidici;
b) RRT = tempo di ritenzione relativo a quello del 4-Fuc; c) Rapporti molari relativi al 3,4-Fuc.
 RRT = tempo di ritenzione relativo a quello del 4-Fuc; c) Rapporti molari relativi al 3,4-Fuc.")
19
DETERMINAZIONE DI SOSTITUENTI NON-SACCARIDICI
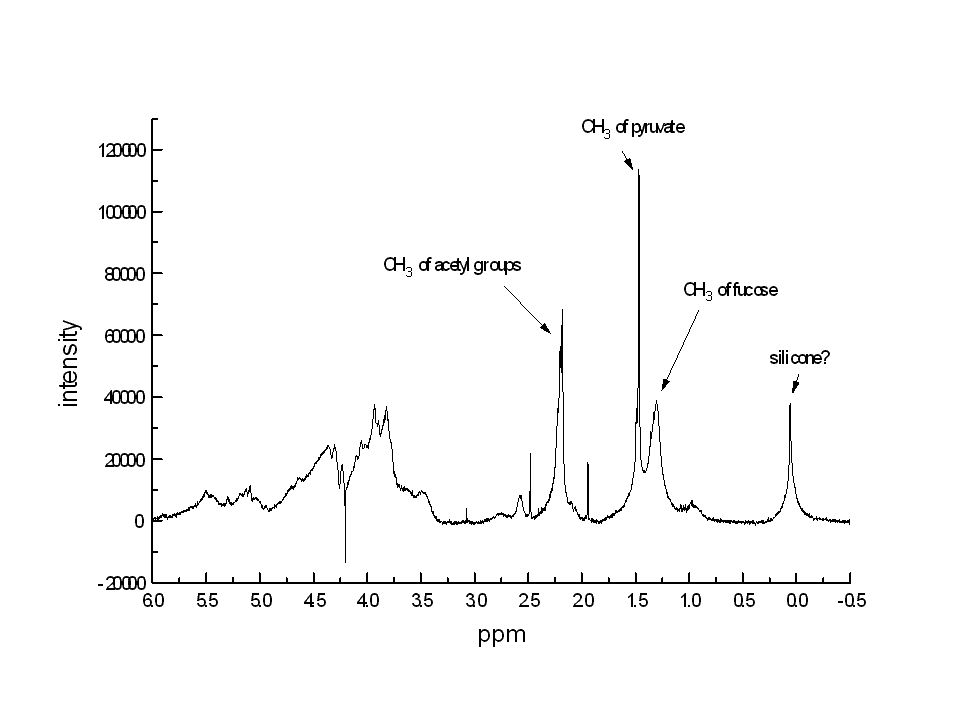
Si ottiene attraverso analisi 1H-NMR poichè le bande di molti di questi sostituenti sono fuori della regione di ppm tipica dei protoni saccaridici. Nel caso del nostro polimero i risultati indicano la presenza di un gruppo piruvile e due gruppi O-acetile per unità ripetitiva del polisaccaride.

22
Il polimero è stato quindi sonicato per diminuire il peso molecolare, e quindi la viscosità della soluzione, che è causa di bande NMR troppo allargate. Polimero in acqua (con acetone a 2.23 ppm) a 70°C piruvato acetone fucosio
 a 70°C. piruvato. acetone. fucosio.")
23
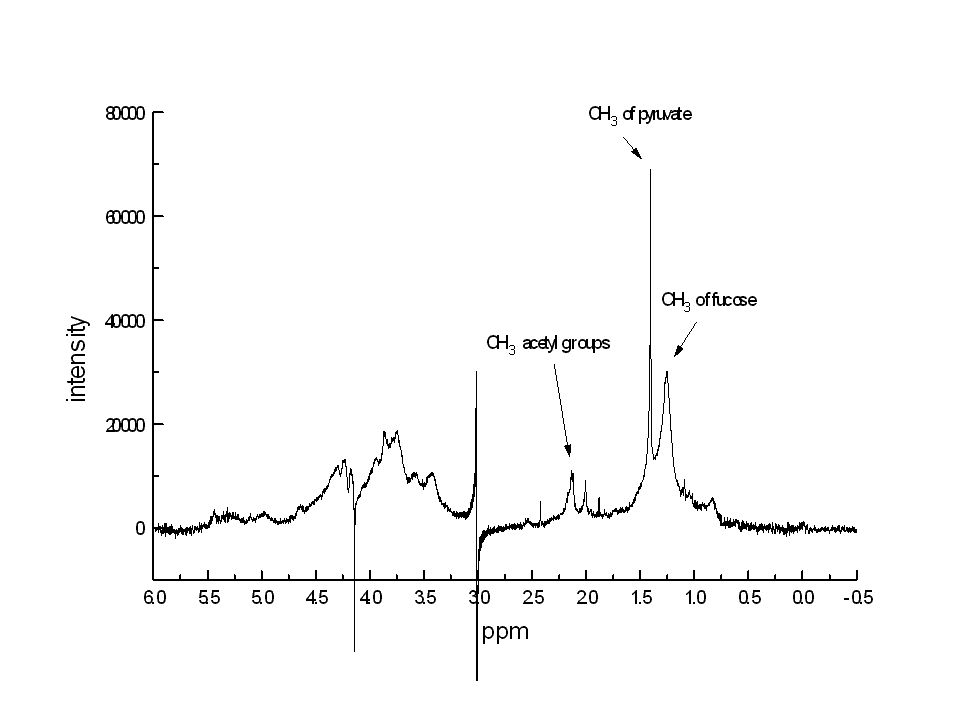
Espansione della zona di risonanza dei protoni anomerici
b c e d f

24
Spesso non è necessario determinare la posizione dei siti di acetilazione e quindi si procede ad una deacetilazione in condizioni basiche che permette anche di semplificare i dati strutturali (soprattutto NMR). Tuttavia, in questo caso, il polimero in soluzione acquosa manteneva ancora alta viscosità che non permettevano di ottenere degli spettri NMR sufficientemente risolti. Per questo motivo è stata effettuata una degradazione di Smith prima dell’analisi NMR.

25
La degradazione di Smith consiste nel rompere il legame C-C in un gruppo diolico. Questo rende il legame glicosidico vicino più labile e quindi si possono ottenere oligomeri che terminano dove era un monosaccaride legato 12 o 14. Per questo è stato utilizzata l’ossidazione con NaIO4 trattando per 7 giorni a 4°C con un rapporto NaIO4/unità ripetitiva dell’EPS = Dopo riduzione dei gruppi aldeidici con NaBH4 il campione è stato trattato con TFA 0,05 M per 7 giorni a temperatura ambiente.

26
La soluzione del polimero degradato è stata poi portata a secco e disciolta in 3 mL di acqua per essere sottoposta a cromatografia di esclusione di volume su una resina P-2 (due colonne in serie) utilizzando acqua come eluente. Una frazione (#47) eluiva come un picco singolo a circa 35h e l’analisi di massa (ESI) ha definito che conteneva un solo componente che è stato quindi sottoposto ad analisi di metilazione ed NMR.
 eluiva come un picco singolo a circa 35h e l’analisi di massa (ESI) ha definito che conteneva un solo componente che è stato quindi sottoposto ad analisi di metilazione ed NMR.")
27
Cromatografia di esclusione di volume dei prodotti della degradazione di Smith

28
La cromatografia di esclusione di volume mostra oltre alla frazione 47 almeno tre picchi sovrapposti vicino al volume escluso e quindi, assumendo che l’idrolisi non era stata completa le tre frazioni sono state raccolte e trattate ancora con TFA 0,05 M per 8 giorni a temp ambiente. La soluzione ottenuta è stata portata a secco e poi ridisciolta in acqua e sottoposta a cromatografia di esclusione di volume. Questa volta il cromatogramma mostrava un picco al volume escluso e due picchi parzialmente sovrapposti vicino. Il più intenso dei due è stato isolato e sottoposto a analisi di massa ESI che ha mostrato la presenza di due oligomeri che sono stati purificati, ripetendo la cromatografia sulle frazioni, e poi sottoposti ad analisi NMR. Questi due composti non saranno discussi concentrando l’attenzione sulla sola frazione 47 la cui parte centrale è stata raccolta ed utilizzata per l’analisi NMR.

29
Spettro di massa ESI della frazione 47
[M+NH4+] = 594.3 [M+Na+] = 599.2 [M+K+] = 615.2 Spettro di massa ESI della frazione 47

30
[3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n
Gli ioni molecolari a m/z 594.3, e (addotti dell’ammonio, del sodio e del potassio) corrispondono ad un oligosaccaride composto di due residui di esoso, un residuo di fucosio e un 4-deossi-treitolo (prodotto della degradazione di Smith che deriva dall’ossidazione del fucosio sostituito in 4 e quindi con la configurazione L). [3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n 3 1 -D-Galp -D-GlcAp 4 -D-Manp CH COOH
![[3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n](http://slideplayer.it/slide/10412373/33/images/30/%5B3%29-%EF%81%A2-D-Glcp-%281%EF%82%AE4%29-%EF%81%A1-L-Fucp-%281%EF%82%AE4%29-%EF%81%A1-L-Fuc-%281%EF%82%AE%5Dn.jpg "Gli ioni molecolari a m/z 594.3, e (addotti dell’ammonio, del sodio e del potassio) corrispondono ad un oligosaccaride composto di due residui di esoso, un residuo di fucosio e un 4-deossi-treitolo (prodotto della degradazione di Smith che deriva dall’ossidazione del fucosio sostituito in 4 e quindi con la configurazione L). [3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n. 3. 1. -D-Galp. -D-GlcAp. 4. -D-Manp CH3 COOH.")
31
Il composto è stato quindi sottoposto a frammentazione durante l’analisi ESI-MS mediante aumento del potenziale sull’orificio di ingresso nello strumento. Assegnazione degli ioni ottenuti dopo frammentazione della frazione 47

32
I frammenti hanno indicato la presenza della sequenza Hex-Fuc-L-4-deossi-treitolo.
Poiché non era presente nessun frammento dimerico Hex-Hex, e considerando che il 4-deossi-treitolo poteva essere originato solo dal fucosio sostutito in 4, si poteva concludere che il secondo residuo di esoso era legato al residuo di fucosio per dare origine ad un tetrasaccaride ramificato.

33
fucosio 3,4-disostituito
L’analisi di metilazione della frazione 47 ha dato come risultato: glucosio terminale galattosio terminale fucosio 3,4-disostituito Con un rapporto molare di 1.24:1.00:0.39. Il basso valore relativo al fucosio 3,4 disostituito può essere associato alla maggior labilità di questo saccaride rispetto agli altri esosi.

34
-D-Glcp-(14)--L-Fucp-(1
3 1 -D-Galp La struttura di questo composto è stata studiata mediante NMR mono- e bi-dimensionale

35
Spettro 1H-NMR della frazione 47
b c Protone anomerico c = Glc Protoni anomerici a = Gal; b = Fuc { { Spettro 1H-NMR della frazione 47

36
Chemical shifts 1H e 13C della frazione 47
Chemical shifts 1H e 13C della frazione 47. I chemical shifts sono dati relativamente al segnale dell’acetone (ref. interno) (2.225 ppm per 1H e ppm per 13C). nd = non determinato
 (2.225 ppm per 1H e ppm per 13C). nd = non determinato.")
37
RIEPILOGO DELLE PRINCIPALI TECNICHE NMR UTILIZZATE NEGLI STUDI STRUTTURALI DI POLISACCARIDI
COSY (Correlation spectroscopy): mette in evidenza nuclei che hanno la stessa costante di accoppiamento TOCSY (Total Correlation Spectroscopy): mette in evidenza nuclei che condividono lo stesso sistema di spin
: mette in evidenza. nuclei che hanno la stessa costante di accoppiamento. TOCSY (Total Correlation Spectroscopy): mette in evidenza nuclei che condividono lo stesso sistema di spin.")
38
HSQC (Heteronuclear Multiple-Quantum Correlation
HSQC (Heteronuclear Multiple-Quantum Correlation Spectroscopy): fornisce una mappa delle connettività (atomi direttamente legati: 1H-13C) HMBC (Heteronuclear Multiple-Bond Correlation Spectoscopy): simile a HSQC, ma mette in evidenza interazioni di spin attraverso più legami (generalmente non superiore a 3 legami) NOESY (Nuclear Overhauser Effect Spectroscopy): mette in evidenza interazioni attraverso lo spazio; adatta a molecole di dimensioni piccole o grandi. ROESY (Rotating frame NOE Spectroscopy): mette in evidenza interazioni attraverso lo spazio; adatta a molecole di medie dimensioni.
: fornisce una mappa delle connettività (atomi direttamente legati: 1H-13C) HMBC (Heteronuclear Multiple-Bond Correlation Spectoscopy): simile a HSQC, ma mette in evidenza interazioni di spin attraverso più legami (generalmente non superiore a 3 legami) NOESY (Nuclear Overhauser Effect Spectroscopy): mette in evidenza interazioni attraverso lo spazio; adatta a molecole di dimensioni piccole o grandi. ROESY (Rotating frame NOE Spectroscopy): mette in evidenza interazioni attraverso lo spazio; adatta a molecole di medie dimensioni.")
39
Spettro 2D-NMR COSY della frazione 47
Fuc 6 Fuc 5 Glc Fuc Gal Spettro 2D-NMR COSY della frazione 47

40
Spettro 2D-NMR TOCSY della frazione 47
Glc 6,6’ 3 5 4 2 Fuc Gal 4 3 2 Spettro 2D-NMR TOCSY della frazione 47

41
Spettro 2D-NMR HSQC della frazione 47
1H1 – 13C1 Glc 1H1 – 13C1 Fuc 1H4 – 13C4 Fuc 1H1 – 13C1 Gal 1H2 – 13C2 Glc Spettro 2D-NMR HSQC della frazione 47

42
Spettro 2D-NMR HMBC della frazione 47
Gal H1 Glc H1 Fuc H1 Fuc C5 Fuc C3 Fuc C4 Spettro 2D-NMR HMBC della frazione 47

43
H1 Glc – H4 Fuc H1 Gal – H3 Fuc Sezione dello spettro ROESY dell’oligosaccaride della frazione 47 registrato a 50°C.

44
[3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n
Dall’analisi di dati analoghi relativi ad altri frammenti e dal paragone dei dati ottenuti sui frammenti con quelli relativi al polimero nativo, si ottiene la struttura definitiva del polisaccaride in studio. [3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n 3 1 -D-Galp -D-GlcAp 4 -D-Manp CH COOH
![[3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n](http://slideplayer.it/slide/10412373/33/images/44/%5B3%29-%EF%81%A2-D-Glcp-%281%EF%82%AE4%29-%EF%81%A1-L-Fucp-%281%EF%82%AE4%29-%EF%81%A1-L-Fuc-%281%EF%82%AE%5Dn.jpg "Dall’analisi di dati analoghi relativi ad altri frammenti e dal paragone dei dati ottenuti sui frammenti con quelli relativi al polimero nativo, si ottiene la struttura definitiva del polisaccaride in studio. [3)--D-Glcp-(14)--L-Fucp-(14)--L-Fuc-(1]n. 3. 1. -D-Galp. -D-GlcAp. 4. -D-Manp CH3 COOH.")
Presentazioni simili


>")


n>")





>")







