Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Effetti sul fenotipo dei diversi tipi di mutazione Mutazioni con perdita di funzione Quasi sempre sono Mutazioni recessive (genotipo in omozigosi): - l’eterozigote non esprime il fenotipo mutante, quindi senza aploinsufficienza (per meccanismi compensatori dell’altro allele?) -con aploinsufficienza (pochissimi i casi di alleli recessivi che esprimono questo fenotipo) Mutazioni con acquisto di funzione In genere sono Mutazioni dominanti - per aumento della produzione proteica (e funzione) rispetto al wt - per produzione di una nuova proteina (e funzione) rispetto al wt
: - l’eterozigote non esprime il fenotipo mutante, quindi senza aploinsufficienza (per meccanismi compensatori dell’altro allele ) -con aploinsufficienza (pochissimi i casi di alleli recessivi che esprimono questo fenotipo) Mutazioni con acquisto di funzione In genere sono Mutazioni dominanti - per aumento della produzione proteica (e funzione) rispetto al wt - per produzione di una nuova proteina (e funzione) rispetto al wt")
2
APPLICAZIONI DELLA TERAPIA GENICA - malattie ereditarie monogeniche con ereditarietà mendeliana (es: fibrosi cistica, SCID, emofilia, talassemia, anemia falciforme) - malattie infettive (es: HIV) -malattie multigeniche (es: tumori del tessuto nervoso, melanomi, tumori ematologici) Terapia genica classica: 1) Produzione e somministrazione della proteina che manca al paziente 2) Eliminazione delle cellule malate 3) Attivazione delle cellule del sistema immunitario che determinano l’uccisione delle cellule malate Terapia genica non convenzionale: Correzione del difetto genetico restaurando la normale espressione genica. 1) Inserimento completo del gene wt nelle cellule somatiche 2) Mutagenesi mirata nelle cellule somatiche per ripristino gene wt 3) Silenziamento nelle cellule somatiche del trascritto del gene mutato 4) Terapia genica nelle cellule germinali (solo in animali modello)
 Inserimento completo del gene wt nelle cellule somatiche 2) Mutagenesi mirata nelle cellule somatiche per ripristino gene wt 3) Silenziamento nelle cellule somatiche del trascritto del gene mutato 4) Terapia genica nelle cellule germinali (solo in animali modello).")
3
Strategie per la terapia genica somatica Aggiunta genica Correzione genica per mutagenesi mirata (gene targeting) Inibizione genica Eliminazione cellulare controllata Eliminazione cellulare mediata dal sistema immunitario Es: profarmaco (retrovirus con gene Tk, Farmaco = ganciclovir)
 Inibizione genica Eliminazione cellulare controllata Eliminazione cellulare mediata dal sistema immunitario Es: profarmaco (retrovirus con gene Tk, Farmaco = ganciclovir)")
4
METODI DI TRASFERIMENTO GENICO 1) Trasferimento genico diretto: - iniezione (con ago) nella cellula di DNA “nudo” o in plasmidi - trasfezione mediata da liposomi (vescicole formate da doppio strato lipidico) 2) Trasferimento tramite vettori virali (trasduzione) tra cui: - retrovirus - adenovirus - virus adeno-associati 3) Tipi di inserzione: - ectopica - mirata
 Trasferimento genico diretto: - iniezione (con ago) nella cellula di DNA nudo o in plasmidi - trasfezione mediata da liposomi (vescicole formate da doppio strato lipidico) 2) Trasferimento tramite vettori virali (trasduzione) tra cui: - retrovirus - adenovirus - virus adeno-associati 3) Tipi di inserzione: - ectopica - mirata")
5
METODI DI TRASFERIMENTO GENICO 1)Trasferimento genico diretto Vantaggi: - Non si generano nuovi virus patogeni - Riduzione del rischio di reazione immunitaria - Possono trasferire molecole di DNA anche molto grandi Svantaggi: - Scarsa efficienza sia di trasferimento che di integrazione - Se integrati, possibile mutagenesi inserzionale
Trasferimento genico diretto Vantaggi: - Non si generano nuovi virus patogeni - Riduzione del rischio di reazione immunitaria - Possono trasferire molecole di DNA anche molto grandi Svantaggi: - Scarsa efficienza sia di trasferimento che di integrazione - Se integrati, possibile mutagenesi inserzionale")
6
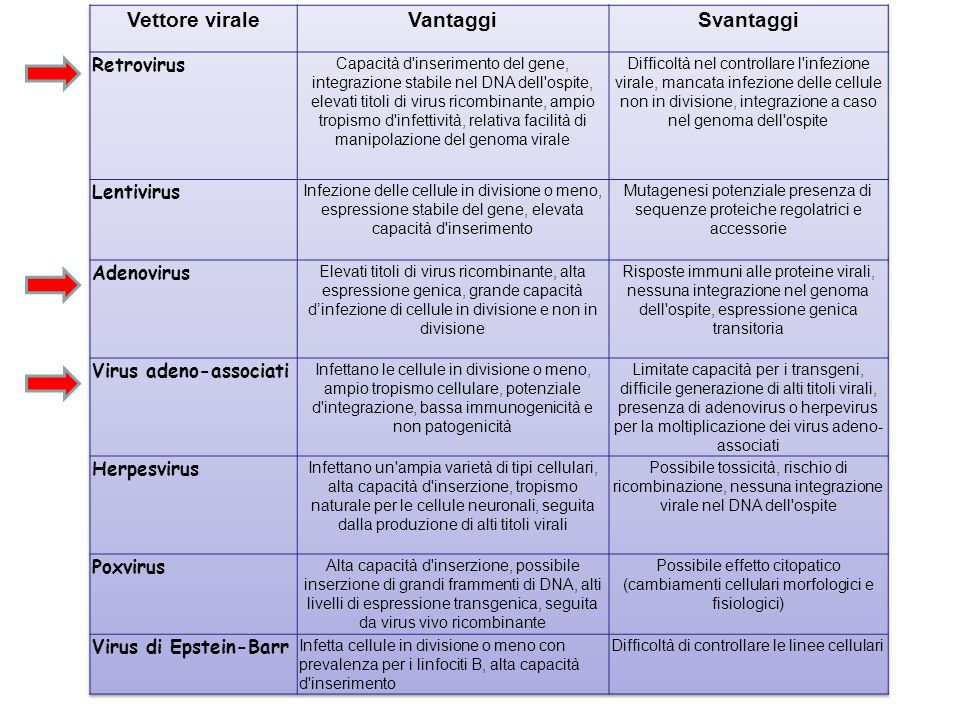
2) Trasferimento tramite vettori virali Caratteristiche necessarie: - Le particelle virali ricombinanti devono essere difettive rispetto alla replicazione (deleti per geni responsabili di replicazione e assemblaggio del virione) e non produrre composti tossici per l’ospite. Vantaggi: - Alta efficienza di trasferimento genico Svantaggi: - Espressione transiente - Possibilità di generare nuovi virus patogeni per ricombinazione con eventuali virus presenti nell’ospite - Inserzione di geni (o molecole di DNA) di dimensioni limitate - Mutagenesi inserzionale (per quelli che si integrano in maniera casuale nel genoma) (attivazione di oncogèni) - Possibilità di reazioni immunitarie - Tecniche con bassa efficienza e costi elevati METODI DI TRASFERIMENTO GENICO
 di dimensioni limitate - Mutagenesi inserzionale (per quelli che si integrano in maniera casuale nel genoma) (attivazione di oncogèni) - Possibilità di reazioni immunitarie - Tecniche con bassa efficienza e costi elevati METODI DI TRASFERIMENTO GENICO.")
7
Terapia genica somatica in vivo ed ex vivo

8
Terapia genica somatica ex vivo - Applicabile solo a certi tipi cellulari: cellule facilmente accessibili; cellule in divisione e coltivabili in vitro (es: da midollo osseo, pelle, tumori);
;")
9
-Estrazione/preparazione delle cellule primarie o della linea cellulare del paziente (cellule autologhe) - Trasferimento del gene in vitro (con o senza vettore virale) - Quando possibile controllo dell’avvenuto inserimento genico - Reimpianto delle cellule nel paziente SVANTAGGI - Applicabile a pochi tessuti (sistema emopoietico, cellule epiteliali, ecc) - Tecnica difficile con scarsa efficienza - Necessità di trattamenti ripetuti nel tempo Esempi: cellule staminali di midollo osseo trattate in emofilici e talassemici Terapia genica somatica ex vivo
 - Trasferimento del gene in vitro (con o senza vettore virale) - Quando possibile controllo dell’avvenuto inserimento genico - Reimpianto delle cellule nel paziente SVANTAGGI - Applicabile a pochi tessuti (sistema emopoietico, cellule epiteliali, ecc) - Tecnica difficile con scarsa efficienza - Necessità di trattamenti ripetuti nel tempo Esempi: cellule staminali di midollo osseo trattate in emofilici e talassemici Terapia genica somatica ex vivo")
10
Introduzione diretta del gene in vivo nelle cellule bersaglio: - iniezione diretta in tipi cellulari non coltivabili in vitro (es. c. cerebrali) o per aumentare il numero di cellule trattate oltre alla terapia ex vivo (es. nel muscolo scheletrico distrofico) - somministrazione in prossimità del tessuto bersaglio (es. attraverso aereosol con adenovirus nelle vie respiratorie nella fibrosi cistica) - il tessuto bersaglio può essere raggiunto in modo sistemico (es: vena porta per raggiungere il fegato, vettori virali a diverso tropismo) Problemi: - Trasferimento quantitativamente limitato - Rischio di inserimento in cellule sane - Reazione immunitaria (alle proteine virali, se si utilizzano virus; talvolta alla proteina espressa) Terapia genica somatica in vivo
 o per aumentare il numero di cellule trattate oltre alla terapia ex vivo (es. nel muscolo scheletrico distrofico) - somministrazione in prossimità del tessuto bersaglio (es. attraverso aereosol con adenovirus nelle vie respiratorie nella fibrosi cistica) - il tessuto bersaglio può essere raggiunto in modo sistemico (es: vena porta per raggiungere il fegato, vettori virali a diverso tropismo) Problemi: - Trasferimento quantitativamente limitato - Rischio di inserimento in cellule sane - Reazione immunitaria (alle proteine virali, se si utilizzano virus; talvolta alla proteina espressa) Terapia genica somatica in vivo.")
11
Terapia genica in vivo per i tumori cerebrali Ganciclovir (Gcv): analogo del nucleoside erpetico: uccide le cellule tk +, ovvero con il vettore virale inserito Gcv Gcv-P Gcv-PPP Incorporazione DNA Terminazione catena rottura DNA Tk HSV Tk mamm VPC = Cellule infettate da Herpes simplex (con gene Tk) che producono virioni impiantate nel tumore Produzione di timidina chinasi virale Somministrazione farmaco (Gcv) Neuronavigazione stereoatassica guidata da MRI
: analogo del nucleoside erpetico: uccide le cellule tk +, ovvero con il vettore virale inserito Gcv Gcv-P Gcv-PPP Incorporazione DNA Terminazione catena rottura DNA Tk HSV Tk mamm VPC = Cellule infettate da Herpes simplex (con gene Tk) che producono virioni impiantate nel tumore Produzione di timidina chinasi virale Somministrazione farmaco (Gcv) Neuronavigazione stereoatassica guidata da MRI")
12
Retrovirus (ad RNA): hanno specifici recettori per entrare nelle cellule; si integrano nei cromosomi in localizzazioni casuali, entrano nel nucleo solo quando si dissolve membrana nucleare (solo cellule in divisione) Adenovirus: legame a specifici recettori cellulari che permettono l'ingresso del virus tramite endocitosi; infettano molti tipi cellulari, anche non in divisione; geni non integrati (episomi); espressione per brevi periodi; necessità di trattamenti ripetuti (es. trattamento fibrosi cistica) Virus adeno-associati (AAV): non sono in grado di replicarsi autonomamente ma necessitano di virus helper co-infettanti come adenovirus o Herpex simplex. Si integrano in sito specifico (19q13.3). TRASFERIMENTO GENICO MEDIANTE VETTORI VIRALI
 Virus adeno-associati (AAV): non sono in grado di replicarsi autonomamente ma necessitano di virus helper co-infettanti come adenovirus o Herpex simplex. Si integrano in sito specifico (19q13.3). TRASFERIMENTO GENICO MEDIANTE VETTORI VIRALI.")
14
Lunga sequenza terminale ripetuta Psi + non codificante: per impacchettamento nel capside Maturazione del genoma a RNA Trascrittasi inversa e integrasi Proteina per involucro Retrovirus Vettore retrovirale Sostituzione di gag, pol, env con gene X: lunghezza max = 8 kb Marcatore selez: gene Neo Inserimento nel vettore a DNA del gene X esogeno e trasfezione di cellule di packaging (HEK293) Lunghezza massima: 8Kb Genoma: 10 Kb costituito da una molecola ssRNA; dopo l’infezione è Retrotrascritto a dsDNA e integrato in ospite Gag = proteine associate genoma; Pol = retrotrascrittasi, integrasi; Env = proteine capside
 Lunghezza massima: 8Kb Genoma: 10 Kb costituito da una molecola ssRNA; dopo l’infezione è Retrotrascritto a dsDNA e integrato in ospite Gag = proteine associate genoma; Pol = retrotrascrittasi, integrasi; Env = proteine capside")
15
Linee cellulari per packaging: forniscono le necessarie funzioni di gag, pol, env, ma delete per gene psi in quanto hanno inserito stabilmente nel genoma i geni virali codificanti per proteine strutturali del capside producono virioni vuoti per l’assemblaggio di virioni completi. Trasfezione transiente con vettore virale ricombinante a DNA. La cellula trascrive i geni del vettore virale producendo mRNA. La transfezione transiente con il vettore ricombinante determina l’impaccamento del costrutto nel virione (contiene l’enzima trascrittasi inversa) vettore virale completo da usare per l’infezione delle cellule da trattare PREPARAZIONE RETROVIRUS PER L’INFEZIONE CELLULARE
 vettore virale completo da usare per l’infezione delle cellule da trattare PREPARAZIONE RETROVIRUS PER L’INFEZIONE CELLULARE.")
16
IL RETROVIRUS NON E’ IN GRADO DA SOLO DI REPLICARSI E PRODURRE PARTICELLE FAGICHE PER ALTRE INFEZIONI CELLULARI INFEZIONE CON RETROVIRUS DELLA CELLULA BERSAGLIO

17
Patogeno naturale dell’uomo, genoma a DNA a doppio filamento, causa infezioni benigne tratto superiore vie respiratorie Genoma di 35-36 kb, dopo delezione fino a 30 kb gene esogeno. Come vettore ha una delezione della regione E1 il che lo rende difettivo per la replicazione. Come cellule d'impaccamento vengono usate cellule renali embrionali (HEK293). Adenovirus (1)
. Adenovirus (1).")
18
L1-L5 proteine strutturali, E1-E4 proteine regolatrici. Come vettore delezione regione E1 che lo rende difettivo per la replicazione. Cellule d'impaccamento: cellule renali embrionali (HEK293). Adenovirus (2)
. Adenovirus (2).")
19
Virus Adenoassociati Appartengono alla famiglia dei parvovirus. Capside icosaedrico e sono privi di un involucro lipidico. Si replicano come DNA a doppio filamento ma si impacchettano per poi infettare come DNA a singolo filamento di circa 5 kb. Per replicarsi hanno bisogno di virus helper (adenovirus o herpes virus). Non sembrano associati a nessuna patologia Ideali come vettori: in assenza di virus helper rimangono integrati in sito specifico (19q13.3); espressione prolungata del transgene in vari tessuti per molti anni. Svantaggi: dimensioni ridotte del transgene (max 4,7 kb)
. Non sembrano associati a nessuna patologia Ideali come vettori: in assenza di virus helper rimangono integrati in sito specifico (19q13.3); espressione prolungata del transgene in vari tessuti per molti anni. Svantaggi: dimensioni ridotte del transgene (max 4,7 kb).")
20
Malattia Gene introdotto Tessuto target ß-talassemia ß-globina Midollo osseo Immunodef. congenita (SCID) ADA Midollo/linfociti Immunodef. Congenita X-linked ILR2G Linfociti Distrofia muscolare Distrofina Muscolo Fibrosi cistica CFTR Epitelio alveoli Emofilia A e B Fattore VIII e IX Epatociti/muscoli Fenilchetonuria Fenilalanina idrossilasi Fegato Morbo di Gaucher Glucocerebrosidasi Midollo osseo Mucopolisaccaridosi tipo I -L-iduronidasi Fibroblasti Mucopolisaccaridosi tipo VII ß-glucuronidasi Midollo osseo Epidermolisi bullosa Laminina Cheratinociti TERAPIA GENICA NELLE MALATTIE UMANE MENDELIANE
 ADA Midollo/linfociti Immunodef. Congenita X-linked ILR2G Linfociti Distrofia muscolare Distrofina Muscolo Fibrosi cistica CFTR Epitelio alveoli Emofilia A e B Fattore VIII e IX Epatociti/muscoli Fenilchetonuria Fenilalanina idrossilasi Fegato Morbo di Gaucher Glucocerebrosidasi Midollo osseo Mucopolisaccaridosi tipo I -L-iduronidasi Fibroblasti Mucopolisaccaridosi tipo VII ß-glucuronidasi Midollo osseo Epidermolisi bullosa Laminina Cheratinociti TERAPIA GENICA NELLE MALATTIE UMANE MENDELIANE.")
21
SCID: SEVERE COMBINED IMMUNODEFICIENCY DISEASE Assenza di Adenosina deaminasi (ADA) (enzima della via di recupero delle purine) Malattia AR: immunodeficienza per mancanza di cellule progenitrici dei linfociti e per accumulo di metaboliti tossici (adenosina e derivati)
 (enzima della via di recupero delle purine) Malattia AR: immunodeficienza per mancanza di cellule progenitrici dei linfociti e per accumulo di metaboliti tossici (adenosina e derivati)")
22
Terapia classica: -Trapianto di midollo: compatibilità del 90-100% dell’HLA (una minoranza dei pazienti) - Somministrazione di enzima ADA estratto dai bovini (correzione del metabolismo, ripristina in parte funzioni immunitarie). Terapia genica ex vivo: - Isolamento delle cellule staminali emopoietiche, oppure - Isolamento dei precursori dei linfociti T - Infezione con vettore retrovirale. Il cDNA dell’ADA dimensioni ridotte (1.5 Kb). Non è necessaria una regolazione fine dell’espressione genica con ripristino della fisiologia sia con alta che bassa efficienza di integrazione - Cellule con copia gene ADA hanno un vantaggio selettivo di crescita eliminando più facilmente i metaboliti tossici - Reinserimento nel midollo del paziente SCID: UN ESEMPIO DI SUCCESSO DELLA TERAPIA GENICA SOMATICA
. Non è necessaria una regolazione fine dell’espressione genica con ripristino della fisiologia sia con alta che bassa efficienza di integrazione - Cellule con copia gene ADA hanno un vantaggio selettivo di crescita eliminando più facilmente i metaboliti tossici - Reinserimento nel midollo del paziente SCID: UN ESEMPIO DI SUCCESSO DELLA TERAPIA GENICA SOMATICA.")
23
Terapia genica per la deficienza di adenosina deaminasi (ADA) Linfociti T oppure Cellule staminali midollo osseo tutte le linee emopoietiche transgeniche
 Linfociti T oppure Cellule staminali midollo osseo tutte le linee emopoietiche transgeniche")
24
Terapia con incremento delle copie geniche ex vivo con retrovirus per la deficienza di adenosina deaminasi (ADA) Terapia 6-7 volte l’anno. Dopo 5 anni recupero parziale della funzione immunitaria ADA Precursori linfociti T da midollo osseo paziente ADA -

25
Human X-Linked Severe Combined Immunodeficiency Mutazioni nel gene IL2RG che codifica le catene gamma del recettore linfocitario per l’interleuchina 2 assenza di espansione e differenziamento dei linfociti T

26
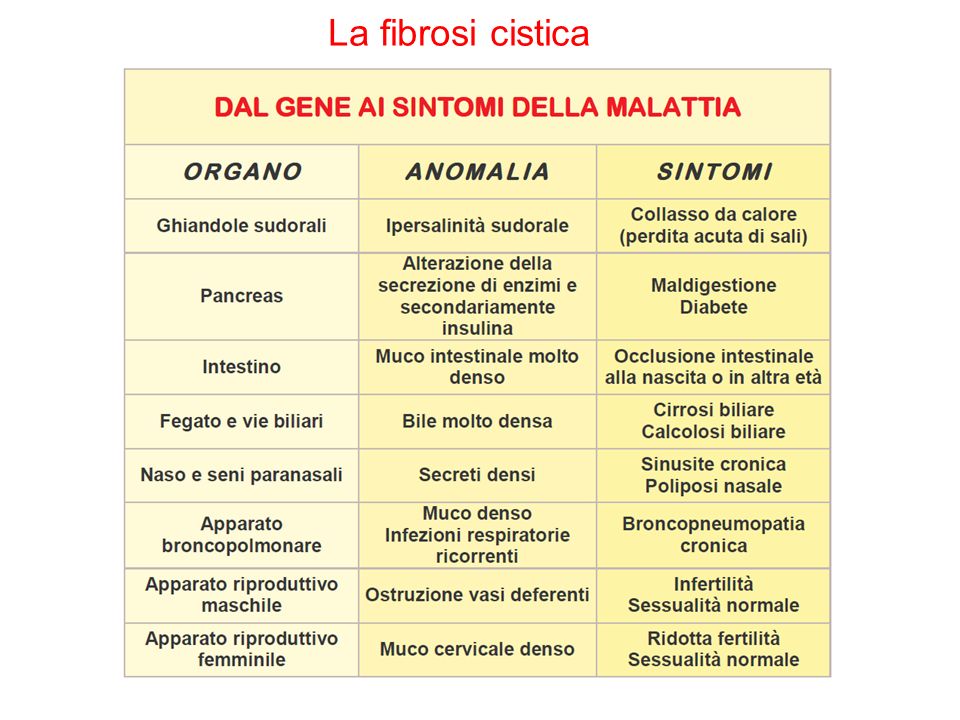
La fibrosi cistica Difetto della proteina CFTR che regola gli scambi elettrolitici, localizzata sulla membrana apicale delle cellule epiteliali Difetto: anomalia del trasporto dei sali: le secrezioni sono «disidratate», sudore ricco di sodio e cloro, muco polmonare denso e vischioso. Malattia letale entro i 30 anni di età senza trattamenti.

27
La fibrosi cistica TEST DEL SUDORE Dosaggio della concentrazione di sale nel sudore Negativo per valori < 40 mEq/L Positivo per valori > 60 mEq/L Non diagnostico per valori intermedi

28
La fibrosi cistica

34
Terapia classica - Drenaggio bronchiale - Somministrazione antibiotici Terapia genica ex vivo - Vettori adenovirali per introdurre il gene CFRT nelle cellule dei polmoni tramite aereosol. - Lentivirus: bassa efficienza trasfezione Problematiche: Tessuto polmonare crea barriere fisiche (muco) e immunitarie; difficile ingresso dell’adenovirus nel nucleo; reazioni infiammatorie; espressione a breve termine Terapia con staminali? Inserimento CFTR in vettori virali e infezione di staminali paziente (da midollo), spinte a differenziarsi ad epiteliali per ripopolare il polmone La fibrosi cistica
 e immunitarie; difficile ingresso dell’adenovirus nel nucleo; reazioni infiammatorie; espressione a breve termine Terapia con staminali. Inserimento CFTR in vettori virali e infezione di staminali paziente (da midollo), spinte a differenziarsi ad epiteliali per ripopolare il polmone La fibrosi cistica.")
35
Le Talassemie Malattie dovute a MUTAZIONI CON PERDITA DI FUNZIONE DEI GENI DELL’EMOGLOBINA

36
Emoglobina (Hb) proteina tetramerica composta da una parte proteica ed una non proteica gruppo eme contiene un atomo di ferro che lega reversibilmente l’ossigeno La parte proteica è costituita da 4 catene polipeptidiche uguali due a due (2 catene di tipo e 2 catene di tipo non- ) Emoglobine embrionali : Hb Gower I ( 2 2 ) Hb Portland ( 2 2 ) Hb Gower II ( 2 2 ) Emoglobina fetale : Hb F ( 2 2 ) Emoglobine adulte : Hb A2 ( 2 2 ) Hb A ( 2 2 )
 proteina tetramerica composta da una parte proteica ed una non proteica gruppo eme contiene un atomo di ferro che lega reversibilmente l’ossigeno La parte proteica è costituita da 4 catene polipeptidiche uguali due a due (2 catene di tipo e 2 catene di tipo non- ) Emoglobine embrionali : Hb Gower I ( 2 2 ) Hb Portland ( 2 2 ) Hb Gower II ( 2 2 ) Emoglobina fetale : Hb F ( 2 2 ) Emoglobine adulte : Hb A2 ( 2 2 ) Hb A ( 2 2 )")
37
Produzione dei vari tipi di catene globiniche nelle diverse fasi della vita di un individuo

38
Le catene di tipo sono lunghe 141 aminoacidi, i loro geni si trovano sul cromosoma 16 (cluster , circa 30 kb) Le catene di tipo sono lunghe 146 aminoacidi, i loro geni si trovano sul cromosoma 11 (cluster , ca. 60 kb) Le catene di tipo e di tipo mostrano una forte omologia di sequenza ad indicare una loro origine evolutiva comune Strachan e Read – Genetica Molecolare Umana, Zanichelli, 2012
 Le catene di tipo e di tipo mostrano una forte omologia di sequenza ad indicare una loro origine evolutiva comune Strachan e Read – Genetica Molecolare Umana, Zanichelli,")
39
alfa talassemie /non- < 1 ( c’è un difetto di catene ) beta talassemie /non- > 1 (c’è un difetto di catene non , che nell’adulto sono sostanzialmente le ) In ogni momento della vita di un individuo (periodo embrionale, fetale e post-natale) il rapporto n° catene /n° catene non- = 1 Si definisce talassemia la condizione in cui tale rapporto 1 Le talassemie sono quindi un difetto QUANTITATIVO generalmente dovuto a perdita di funzione dei geni che codificano le globine
 beta talassemie /non- > 1 (c’è un difetto di catene non , che nell’adulto sono sostanzialmente le ) In ogni momento della vita di un individuo (periodo embrionale, fetale e post-natale) il rapporto n° catene /n° catene non- = 1 Si definisce talassemia la condizione in cui tale rapporto 1 Le talassemie sono quindi un difetto QUANTITATIVO generalmente dovuto a perdita di funzione dei geni che codificano le globine")
40
Microcitemia o Talassemia minor o Trait talassemico sono sinonimi e indicano la condizione CLINICAMENTE ASINTOMATICA degli eterozigoti facilmente individuabili attraverso un semplice esame ematologico Anemia mediterranea o Morbo di Cooley o Talassemia major sono sinonimi e indicano il quadro clinico, molto grave (mortale in assenza di cure) degli individui omozigoti (o eterozigoti composti per alleli non funzionanti) alleli b thal0 (a thal0 ) causano assenza completa di catene beta (alfa) alleli b thal+ (a thal+ ) causano una riduzione più o meno marcata di catene beta (alfa)
 degli individui omozigoti (o eterozigoti composti per alleli non funzionanti) alleli b thal0 (a thal0 ) causano assenza completa di catene beta (alfa) alleli b thal+ (a thal+ ) causano una riduzione più o meno marcata di catene beta (alfa)")
41
NEI MALATI NON CURATI SI OSSERVANO GRAVISSIME DEFORMAZIONI DEL CRANIO E DELLA FACCIA, OLTRE A EPATO- E SPLENOMEGALIA Anemia mediterranea o Morbo di Cooley o Talassemia major

42
Microcitemia o Talassemia minor o Trait talassemico Nei microcitemici la quantità totale di Hb è ridotta mentre il numero di globuli rossi è aumentato Gli eritrociti sono però più piccoli Il ridotto contenuto di Hb determina un appiattimento delle emazie e una loro maggiore resistenza all’emolisi in soluzione salina ipotonica.

43
Schema riassuntivo della patofisiologia delle beta talassemie

44
FREQUENZA DELLA MICROCITEMIA NEL MONDO Portatori sani nel mondo circa 180.000.000; in assenza di prevenzione le nascite di nuovi malati sarebbero 70.000 ogni anno In Italia vivono circa 2.500.000 microcitemici sani Grazie alla prevenzione le nascite di individui malati sono molto limitate

45
Terapie per le Talassemie

46
COSA È LA DMD LA DISTROFIA MUSCOLARE DI DUCHENNE (DMD) E’ UNA GRAVE PATOLOGIE GENETICA, CAUSATA DALLA TOTALE ASSENZA DELLA DISTROFINA NEL MUSCOLO STRIATO E LISCIO. colpisce tutti i muscoli scheletrici, la muscolatura liscia e il cuore. difficoltà di deambulazione e di movimento insufficienza respiratoria problemi cardiaci DMD: conduce alla completa immobilità, l’aspettativa di vita non supera i 25-30 anni. A 10-12 anni i ragazzi vivono sulla carrozzina. La DMD è una malattia rara, con una ha una frequenza di 1 su 3500 Mutazioni dello stesso gene causano anche: La distrofia di Becker (DMB), meno invalidante con assenza parziale di distrofina. Cardiomiopatia dilatativa legata all’X
, meno invalidante con assenza parziale di distrofina. Cardiomiopatia dilatativa legata all’X.")
47
Il gene è localizzato sul braccio corto del cromosoma X. Le donne con una sola copia mutata del gene sono portatrici sane, gli uomini, essendo emizigoti, si ammalano. E’ il più lungo gene fin ora identificato, circa 2,3 Mb (79 esoni) mRNA rappresenta solo lo 0,5% del gene (circa 12 kb) 7 promotori, originano 1 proteina lunga e 6 versioni più corte. La distrofina, formata da 3685 aminoacidi, PM 427 kDa. TUTTA COLPA DI UN GENE
 mRNA rappresenta solo lo 0,5% del gene (circa 12 kb) 7 promotori, originano 1 proteina lunga e 6 versioni più corte. La distrofina, formata da 3685 aminoacidi, PM 427 kDa. TUTTA COLPA DI UN GENE.")
48
LA DISTROFINA Ancorata sulla faccia interna della membrana delle fibre muscolari. C- terminale è legato ad altre proteine di membrana. N-terminale è connesso alle strutture contrattili all’interno della cellula muscolare. Determinante per la stabilità meccanica della membrana durante la contrazione muscolare, funziona da ammortizzatore. Assenza o malfunzionamento causa rottura della membrana muscolare: Ingresso del calcio nelle fibre. Attivazione di enzimi. Infiammazione e attivazione dei fibroblasti. Formazione di tessuto cicatriziale che sostituisce quello muscolare. Degenerazione muscolare e insorgenza della patologia.

49
Terapia genica per la DMD - Terapia di tipo sistemico (muscolatura striata, cuore, diaframma) - Muscoli target difficilmente raggiungibili - Transgene di dimensioni elevate (cDNA di 14 kb) - Pochi vettori utilizzabili
 - Muscoli target difficilmente raggiungibili - Transgene di dimensioni elevate (cDNA di 14 kb) - Pochi vettori utilizzabili")
50
TERAPIA GENICA: L’EXON SKIPPING La mutazione elimina EX50 portando alla fusione di IVS 49 e IVS50; cambia il reading frame del gene della distrofina e cessa la produzione della proteina funzionale Viene indotto l’exon skipping (E.S.) dell’esone 51 nel pre-mRNA: Antisense Oligo (AO) che si attaccano alla sequenza dell’ESE (exonic-splicing-enhancer) all’interno dell’esone 51 E.S. ristabilisce il corretto schema di lettura (reading frame) modificando l’mRNA (EX49 in frame con EX52) La proteina è più corta ma ancora funzionante Converte la DMD in DMB in modo da ridurre la gravità della malattia
 modificando l’mRNA (EX49 in frame con EX52) La proteina è più corta ma ancora funzionante Converte la DMD in DMB in modo da ridurre la gravità della malattia.")
51
COME FUNZIONA L’EXON SKIPPING 1/2 AOS o Oligonucleotidi antisenso, corti frammenti di RNA, si appaiano in regioni coinvolte nello splicing del pre –mRNA, producendo splicing alternativo con rimozione di uno o piu esoni i 2 tipi di AOS più utilizzati nella sperimentazione per l’eliminazione dell’esone 51 sono: 2’O- metyl- fosforotioates, PRO05, formato da 20 basi Morfolino, AVI-4658, ha 30 basi, e include le 20 del PRO05 2’O-Metil AO morfolino AO UCUUUACGGUGAAGGAACUGAUCUUUACGGUGAAGGAACUACAACCYC

52
COME FUNZIONA L’EXON SKIPPING 2/2 Gli AOs si attaccano alla sequenza dell’ESE (exonic-splicing-enhancer) all’interno dell’esone 51. ESE sequence è importante per un normale processo di splicing, ma se è bloccata da un AO, l’esone viene eliminato con le sequenze introniche.

53
Il trattamento con una molecola antisenso, somministrata con iniezione intramuscolare. Lo studio, compiuto presso UCL (Londra) ha riguardato 7 ragazzi di età compresa tra 10 e 17 anni con DMD a cui era stata diagnosticata la malattia e in cui poteva risultare di beneficio lo skipping (salto) dell’esone 51. Due dei pazienti hanno ricevuto 0.09 mg dell’oligonucleotide antisenso AVI-4658, iniettato localmente in un piccolo muscolo del piede (extensor digitorum brevis). Gli altri 5 pazienti hanno ricevuto 0.9 mg di AVI-4658 nell’altro piede. I pazienti che hanno ricevuto il più basso dosaggio (0.09 mg) di AVI-4658 hanno mostrato poca espressione di distrofina, il dosaggio più alto (0.9 mg) ha prodotto un aumento dell’espressione di distrofina nel muscoli trattati. Nelle aree delle sezioni immunocolorate adiacenti al luogo in cui AVI-4658 è stato iniettato, il 44-79% delle miofibre ha aumentato l’espressione della distrofina. Fonte: Lancet Neurology, 2009
 ha riguardato 7 ragazzi di età compresa tra 10 e 17 anni con DMD a cui era stata diagnosticata la malattia e in cui poteva risultare di beneficio lo skipping (salto) dell’esone 51. Due dei pazienti hanno ricevuto 0.09 mg dell’oligonucleotide antisenso AVI-4658, iniettato localmente in un piccolo muscolo del piede (extensor digitorum brevis). Gli altri 5 pazienti hanno ricevuto 0.9 mg di AVI-4658 nell’altro piede. I pazienti che hanno ricevuto il più basso dosaggio (0.09 mg) di AVI-4658 hanno mostrato poca espressione di distrofina, il dosaggio più alto (0.9 mg) ha prodotto un aumento dell’espressione di distrofina nel muscoli trattati. Nelle aree delle sezioni immunocolorate adiacenti al luogo in cui AVI-4658 è stato iniettato, il 44-79% delle miofibre ha aumentato l’espressione della distrofina. Fonte: Lancet Neurology,")
54
PROTOCOLLI PER SPERIMENTAZIONI CLINICHE DI TERAPIE GENICHE Protocolli approvati per terapia genica: per quasi 2/3 riguardano il cancro Utilizzo di vettori per i protocolli attuali Terapia genica nei tumori Se inattivato gene soppressore tumore (oncosoppressore) introduzione nuova versione gene; Se attivato oncogène spegnimento con gene per RNA antisenso
 introduzione nuova versione gene; Se attivato oncogène spegnimento con gene per RNA antisenso")
55
Strategie di terapia genica somatica in malattie da acquisizione di funzione e per spegnere oncogèni Produzione di RNA interferenti introdotti con vettori virali

57
RISC: RNA-Induced Silencing Complex. (Formato da RNA e proteine) srotola ds RNA e il filamento complementare si appaia con RNA da silenziare, degradazione dell’ RNA a doppio filamento Complesso Dicer: RNAsi che degrada lunghi filamenti di dsRNA a frammenti di circa 20 bp Si introduce nella cellula l’RNA del gene che si vuole silenziare a doppio filamento (dsRNA)
 srotola ds RNA e il filamento complementare si appaia con RNA da silenziare, degradazione dell’ RNA a doppio filamento Complesso Dicer: RNAsi che degrada lunghi filamenti di dsRNA a frammenti di circa 20 bp Si introduce nella cellula l’RNA del gene che si vuole silenziare a doppio filamento (dsRNA).")
58
Es: ganciclovir Terapia genica dei tumori Gcv Gcv-P Gcv-PPP Incorporazione DNA Terminazione catena rottura DNA Tk HSV Tk mamm Ganciclovir (Gcv): analogo di un nucleoside virale (metilguanina): uccide le cellule tk +, ovvero quelle con il vettore virale inserito
: analogo di un nucleoside virale (metilguanina): uccide le cellule tk +, ovvero quelle con il vettore virale inserito")
59
Caratteristiche del vettore ideale per la TERAPIA GENICA Efficienza (trasduzione di un numero di cellule elevato). Garanzia di una prolungata produzione della proteina terapeutica (a livelli adeguati). Capacità di incorporare DNA di varie dimensioni. Garanzia nella regolazione (trascrizionale, traduzionale o post-traduzionale) dell’espressione del gene introdotto Non patogenicità Facilità di somministrazione al paziente. Capacità di raggiungere specificamente le cellule bersaglio. Buona tolleranza. Assenza di immunogenicità Facilità di costruzione e riproducibilità IL VETTORE IDEALE NON ESISTE. I VETTORI PIU’ UTILIZZATI OGGI SONO RETROVIRUS E LENTIVIRUS
. Capacità di incorporare DNA di varie dimensioni. Garanzia nella regolazione (trascrizionale, traduzionale o post-traduzionale) dell’espressione del gene introdotto Non patogenicità Facilità di somministrazione al paziente. Capacità di raggiungere specificamente le cellule bersaglio. Buona tolleranza. Assenza di immunogenicità Facilità di costruzione e riproducibilità IL VETTORE IDEALE NON ESISTE. I VETTORI PIU’ UTILIZZATI OGGI SONO RETROVIRUS E LENTIVIRUS.")
Presentazioni simili
























 Trasferimento genico tramite microiniezione nel pronucleo maschile di una cellula uovo appena fecondata. Se l’inserimento.>")