Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Da un caso di neonata con familiarità per
Journal Club Siena, 19 Febbraio 2013 Da un caso di neonata con familiarità per CITRULLINEMIA II: I DIFETTI DEL CICLO DELL’UREA Dott.ssa Silvia Franchi Scuola di Specializzazione in Pediatria

2
Lo spunto di riflessione deriva dal caso di una neonata gemella di EG pari a 28+5 settimane (ricoverata in TIN e quindi in PIN) con familiarità per malattia metabolica (CITRULLINEMIA II) cugina di primo grado nella linea materna (nata nel 2008 da genitori non consanguinei, parto eutocico a 40+5 settimane di EG. Valori auxologici alla nascita e perinatalità nella norma). Allo screening neonatale: aumento dei valori di Citrullinemia (152 umoli/l) Ulteriore aumento a 13 giorni di vita (350 umoli/l). Lieve incremento delle transaminasi Ammoniemia nella norma Acido Orotico urinario nella norma. Contatto con U.O. di malattie Metaboliche (Ospedale Meyer): La Prof.ssa Donati ci consiglia di controllare continuamente le TRANSAMINASI e, in caso di aumento dei valori oltre il range di normalità, eseguire AMMONIEMIA ed AMINOACIDOGRAMMA nel sospetto di una anomalia del ciclo dell’Urea… CITRULLINEMIA: valori normali 10-20 micromoli/l
 Ulteriore aumento a 13 giorni di vita (350 umoli/l). Lieve incremento delle transaminasi. Ammoniemia nella norma. Acido Orotico urinario nella norma. Contatto con U.O. di malattie Metaboliche (Ospedale Meyer): La Prof.ssa Donati ci consiglia di controllare continuamente le TRANSAMINASI e, in caso di aumento dei valori oltre il range di normalità, eseguire AMMONIEMIA ed AMINOACIDOGRAMMA nel sospetto di una anomalia del ciclo dell’Urea… CITRULLINEMIA: valori normali micromoli/l.")
3
Catabolismo degli aminoacidi
Formazione di Ammonio libero (altamente tossico per il SNC!) Detossificazione epatica ad urea, espulsa con le urine (TRAMITE IL CICLO DELL’UREA o dell’Ornitina)
 Detossificazione epatica ad urea, espulsa con le urine. (TRAMITE IL CICLO DELL’UREA o dell’Ornitina)")
4
Acido Argininosuccinico
CPS Ciclo dell’Urea OCT NH3 + bicarbonato Carbamil-P Ornitina Citrullina ASS UREA ARGINASI Ac.Aspartico Arginina Acido Argininosuccinico Acido Fumarico AL ENZIMI COINVOLTI Carbamilfosfato sintetasi (CPS), Ornitina carbamil-transferasi (OCT), Argininsuccinato sintetasi (AS), Argininsuccinato liasi (AL) Arginasi Un sesto enzima, N-acetilglutammato sintetasi (NAGs), è richiesto per la sintesi di N-acetilglutammato, il quale è un attivatore dell'enzima carbamilfosfato sintetasi (CPS). Nelson, Textbook of Pediatrics, XVI Edition
, Ornitina carbamil-transferasi (OCT), Argininsuccinato sintetasi (AS), Argininsuccinato liasi (AL) Arginasi. Un sesto enzima, N-acetilglutammato sintetasi (NAGs), è richiesto per la sintesi di N-acetilglutammato, il quale è un attivatore dell enzima carbamilfosfato sintetasi (CPS). Nelson, Textbook of Pediatrics, XVI Edition.")
5
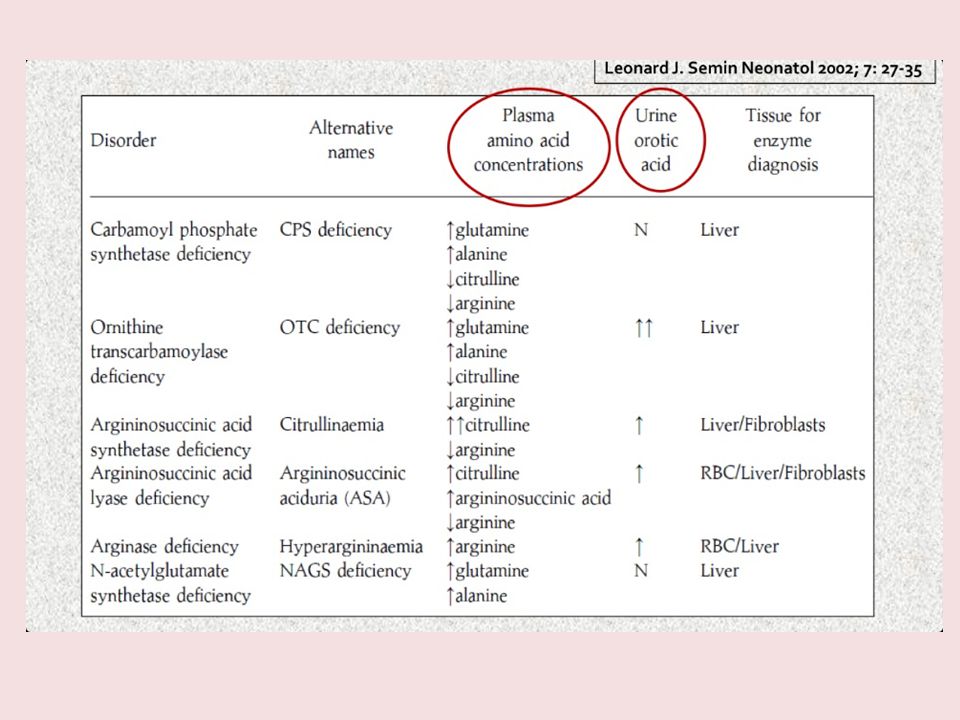
Origine genetica dei principali difetti enzimatici
Il problema è che… Sono stati osservati deficit isolati di tali enzimi (DEFICIT ENZIMATICI CONGENITI) con prevalenza totale di 1 su nati vivi e sono le più frequenti cause genetiche di iperammoniemia nel bambino (Nelson, Textbook of Pediatrics, XVI Edition). Mentre ciascun quadro è contraddistinto da un accumulo di differenti precursori, l’iperammoniemia e l’iperglutamminemia sono caratteristiche comuni a tutti i disordini (Roth KS(2007) Hyperammonaemia (Accessed: 29 December 2007: Last updated 31 May 2007) Essi possono essere distinti l'uno dall'altro generalmente solo attraverso esami di laboratorio, a meno che non si sospetti una trasmissione X-linked.. CPS : AR 2q OTC: X-linked Xp21 ASS: AR 9q ASL: AR 7q ARG: AR 6q NAGs: AR 17q Origine genetica dei principali difetti enzimatici
 con prevalenza totale di 1 su nati vivi e sono le più frequenti cause genetiche di iperammoniemia nel bambino. (Nelson, Textbook of Pediatrics, XVI Edition). Mentre ciascun quadro è contraddistinto da un accumulo di differenti precursori, l’iperammoniemia e l’iperglutamminemia sono caratteristiche comuni a tutti i disordini. (Roth KS(2007) Hyperammonaemia (Accessed: 29 December 2007: Last updated 31 May 2007) Essi possono essere distinti l uno dall altro generalmente solo attraverso esami di laboratorio, a meno che non si sospetti una trasmissione X-linked.. CPS : AR 2q. OTC: X-linked Xp21. ASS: AR 9q. ASL: AR 7q. ARG: AR 6q. NAGs: AR 17q. Origine genetica dei principali difetti enzimatici.")
6
Insorgenza neonatale In generale:
A causa della drammatica presentazione clinica di queste malattie nel periodo neonatale e a causa delle conseguenze a lungo termine dell'iperammoniemia in tale epoca, è fondamentale sottolineare che i deficit enzimatici possono presentarsi Nel periodo neonatale Tardivamente (anche in età adulta) quadro sfumato Insorgenza neonatale Il bambino, quasi sempre nato al termine di una gravidanza normale, senza alcun fattore di rischio prenatale o perinatale, con travaglio e parto normali, appare sano per almeno 24 ore. Tra le 24 e le 72 ore (occasionalmente molti giorni dopo) diventa letargico e necessita di stimoli per alimentarsi. Nel giro di ore possono comparire vomito, letargia ingravescente ed ipotermia. Viene spesso presa in considerazione la diagnosi di emorragia intracranica, se si nota una fontanella sporgente od un aumento di volume della testa. La TC cerebrale rivela la presenza di edema. Se non è misurato il livello di ammonio plasmatico, la morte del neonato può essere attribuita a sepsi o emorragia intracranica. EEG : onde lente alcalosi iperammoniemia
 quadro sfumato. Insorgenza neonatale. Il bambino, quasi sempre nato al termine di una gravidanza normale, senza alcun fattore di rischio prenatale o perinatale, con travaglio e parto normali, appare sano per almeno 24 ore. Tra le 24 e le 72 ore (occasionalmente molti giorni dopo) diventa letargico e necessita di stimoli per alimentarsi. Nel giro di ore possono comparire vomito, letargia ingravescente ed ipotermia. Viene spesso presa in considerazione la diagnosi di emorragia intracranica, se si nota una fontanella sporgente od un aumento di volume della testa. La TC cerebrale rivela la presenza di edema. Se non è misurato il livello di ammonio plasmatico, la morte del neonato può essere attribuita a sepsi o emorragia intracranica. EEG : onde lente. alcalosi. iperammoniemia.")
7
Presentazione clinica tardiva (nel bambino e nell’adulto)
Iporessia cronica rifiuto alimenti proteici malnutrizione disturbi del sonno alterazioni comportamentali allucinazioni visive scotomi psicosi atassia Crisi di scompenso metabolico slatentizzate da episodi infettivi intercorrenti o aumentato apporto proteico. Nel corso delle crisi : vomito, cefalea, sopore coma, iperammoniemia e alterazione della funzione epatica. EEG: onde lente TAC, RM :edema cerebrale nelle fasi acute; atrofia cerebrale

8
Alcuni esempi… partendo dal nostro caso
Citrullinemia tipo 1: AR, legata a deficit di ARGININO-SUCCINICO SINTETASI (ASS). Esordio: nei primi giorni di vita improvvisamente Esito: potenzialmente infausto (COMA IPERAMMONIEMICO e ACIDOSI LATTICA). A volte: i sintomi sono più tardivi e meno evidenti ma si palesano con anoressia, vomito, ipotonia, ritardo della crescita, ritardo psicomotorio e convulsioni. Diagnosi: Prenatale se sospetto per familiarità (amniocentesi) o Postnatale: AMMONIEMIA (AUMENTATA); aumento di citrullina ( microMoli: vn 10-20), glutammina e dell'alanina, riduzione di arginina e aciduria orotica. La terapia alimentare dell'ipercitrullinemia è ipo-proteica coadiuvata a supplementazione di arginina, benzoato e fenilbutirrato di sodio (detossificazione). Citrullinemia tipo 2 ESORDIO VARIABILE, ANCHE TARDIVO, IN ETA’ ADULTA. Può portare ritardo mentale di grado lieve e insufficienza epatica.
. Esordio: nei primi giorni di vita improvvisamente. Esito: potenzialmente infausto (COMA IPERAMMONIEMICO e ACIDOSI LATTICA). A volte: i sintomi sono più tardivi e meno evidenti ma si palesano con anoressia, vomito, ipotonia, ritardo della crescita, ritardo psicomotorio e convulsioni. Diagnosi: Prenatale se sospetto per familiarità (amniocentesi) o Postnatale: AMMONIEMIA (AUMENTATA); aumento di citrullina ( microMoli: vn 10-20), glutammina e dell alanina, riduzione di arginina e aciduria orotica. La terapia alimentare dell ipercitrullinemia è ipo-proteica coadiuvata a supplementazione di arginina, benzoato e fenilbutirrato di sodio (detossificazione). Citrullinemia tipo 2 ESORDIO VARIABILE, ANCHE TARDIVO, IN ETA’ ADULTA. Può portare ritardo mentale di grado lieve e insufficienza epatica.")
9
Citrullinemia II A.R. “Type II citrullinemia is primarily found in the Japanese population, where it occurs in an estimated one in 100,000 to 230,000 individuals. Type II has also been reported in people from East Asian and Middle Eastern populations. Mutations in the SLC25A13 gene are responsible for type II citrullinemia. This gene makes a protein called citrin, which normally shuttles certain molecules in and out of mitochondria. These molecules are essential for the urea cycle and are also involved in making proteins and nucleotides. Mutations in SLC25A13 typically prevent the production of any functional citrin, which inhibits the urea cycle and disrupts the production of proteins and nucleotides. The resulting buildup of ammonia and other toxic substances leads to the symptoms that usually appear during adulthood and mainly affect the central nervous system. Characteristic features include confusion, abnormal behaviors (such as aggression, irritability, and hyperactivity), seizures, and coma. These symptoms can be triggered by certain medications, infections, and alcohol intake in people with this type. Researchers have found many infants with neonatal intrahepatic cholestasis have the same mutations in the SLC25A13 gene as adults with type II citrullinemia.” May also develop in people who had neonatal cholestasis during infancy. This condition blocks the flow of bile and prevents the body from processing certain nutrients properly. In many cases, the symptoms resolve within a year. Years or even decades later, however, some of these people develop the characteristic features of adult type II citrullinemia. (Online 'Mendelian Inheritance in Man' (OMIM) and Online 'Mendelian Inheritance in Man' (OMIM) )
, seizures, and coma. These symptoms can be triggered by certain medications, infections, and alcohol intake in people with this type. Researchers have found many infants with neonatal intrahepatic cholestasis have the same mutations in the SLC25A13 gene as adults with type II citrullinemia. May also develop in people who had neonatal cholestasis during infancy. This condition blocks the flow of bile and prevents the body from processing certain nutrients properly. In many cases, the symptoms resolve within a year. Years or even decades later, however, some of these people develop the characteristic features of adult type II citrullinemia. (Online Mendelian Inheritance in Man (OMIM) and Online Mendelian Inheritance in Man (OMIM) )")
10
Citrullinemia II (OMIM)
“The neonatal intrahepatic cholestasis develops between one to five months of age. The adult onset years of age. Neonatal intrahepatic cholestasis caused by citrin deficiency has been diagnosed in over 70 infants between one to five months of age. In addition to intrahepatic cholestasis, they have jaundice and fatty liver at biopsy. Three patients developed liver failure necessitating transplants before 12 months of age. Symptoms may be acute or develop gradually and include enuresis, delayed menarche, insomnia, nocturnal sweats and terrors, recurrent vomiting,diarrhea, tremors, confusion, lethargy, convulsions, delusions, hallucinations and episodes of coma. Hypercitrullinemia and hyperammonemia are present. Pancreatitis, hyperlipidemia or death from cerebral edema generally occurs within a few years of the diagnosis. Hepatocellular carcinoma has been reported in a few cases. Physical Findings: No dysmorphisms. Treatment: Liver transplant in the adult form, it is not known if the neonatal form patients will go on to develop the adult form. Neonatal symptoms tend to resolve with protein restriction. Arginine may help ameliorate the symptoms. Natural History without treatment: The neonatal form may resolve. The adult form progresses to death.

11
ALTRI DIFETTI DEL CICLO DELL’UREA…
Deficit di NAG sintetasi: variabilità fenotipica iperammoniemia grave, iperammoniemia lieve associata paradossalmente ad encefalopatia grave, diarrea ricorrente ed acidosi, disturbi del movimento, ipoglicemia, iperornitinemia, livelli plasmatici di arginina e citrullina normali, così come il contenuto epatico di NAG. La diagnosi si basa sul saggio dell'attività dell'enzima epatico. Il deficit di AL ha due caratteristiche distintive: epatomegalia grave, nella forma ad insorgenza precoce, ed alterazioni dei capelli (tricoressi nodosa) nella forma ad insorgenza tardiva (alterazioni dei capelli simili sono state descritte anche nel deficit di AS). DEFICIT DI ARGINASI Sintomi principali, tutti progressivamente ingravescenti: tetraplegia spastica, che colpisce di più gli arti inferiori rispetto ai superiori, crisi epilettiche, ritardo psicomotorio, iperattività, difficoltà di crescita, e talvolta atetosi. Può verificarsi che l'iperammoniemia sintomatica progredisca verso l'encefalopatia (i livelli plasmatici di ammonio sono da tre a quattro volte i valori normali, raramente anche sei volte i valori normali) . Nonostante esista variabilità fenotipica, con alcuni casi presumibilmente asintomatici a quattro anni di età, un'accurata analisi dei casi riportati suggerisce che le manifestazioni cliniche si verifichino precocemente durante il primo anno di vita; esse comprendono irritabilità, pianto inconsolabile, anoressia, vomito, e tappe di sviluppo acquisite con ritardo. (C. Scriver et al., The Metabolic and Molecular Bases of Inherited Disease, Eighth Edition).
 nella forma ad insorgenza tardiva (alterazioni dei capelli simili sono state descritte anche nel deficit di AS). DEFICIT DI ARGINASI. Sintomi principali, tutti progressivamente ingravescenti: tetraplegia spastica, che colpisce di più gli arti inferiori rispetto ai superiori, crisi epilettiche, ritardo psicomotorio, iperattività, difficoltà di crescita, e talvolta atetosi. Può verificarsi che l iperammoniemia sintomatica progredisca verso l encefalopatia (i livelli plasmatici di ammonio sono da tre a quattro volte i valori normali, raramente anche sei volte i valori normali) . Nonostante esista variabilità fenotipica, con alcuni casi presumibilmente asintomatici a quattro anni di età, un accurata analisi dei casi riportati suggerisce che le manifestazioni cliniche si verifichino precocemente durante il primo anno di vita; esse comprendono irritabilità, pianto inconsolabile, anoressia, vomito, e tappe di sviluppo acquisite con ritardo. (C. Scriver et al., The Metabolic and Molecular Bases of Inherited Disease, Eighth Edition).")
12
DEFICIT di OCT (X-linked dominante o recessiva)
Deficit enzimatico completo: si manifesta con coma iperammoniemico neonatale molto grave, di solito letale. Nelle eterozigoti è asintomatico oppure provoca una escrezione di acido orotico (spontaneamente o dopo carico proteico), che permette di riconoscere le portatrici, oppure determina una malattia sintomatica di gravità variabile: disgusto per le proteine fino al vomito ciclico ritardo della crescita ipotonia ritardo psicomotorio episodi di coma iperammoniemico patologie psichiatriche. Nelle donne, la gravità della malattia è variabile in funzione del grado di inattivazione del cromosoma X che porta la mutazione. Un’attività enzimatica residua causa coma iperammoniemico, ad esordio tardivo, simile alla sindrome di Reye o encefalite, a volte diagnosticato solo tardivamente, in età adulta.
, che permette di riconoscere le portatrici, oppure determina una malattia sintomatica di gravità variabile: disgusto per le proteine fino al vomito ciclico. ritardo della crescita. ipotonia. ritardo psicomotorio. episodi di coma iperammoniemico. patologie psichiatriche. Nelle donne, la gravità della malattia è variabile in funzione del grado di inattivazione del cromosoma X che porta la mutazione. Un’attività enzimatica residua causa coma iperammoniemico, ad esordio tardivo, simile alla sindrome di Reye o encefalite, a volte diagnosticato solo tardivamente, in età adulta.")
14
Quando si dovrebbe sospettare un difetto ereditario del metabolismo?
In ogni neonato che presenti: deterioramento neurologico immotivato letargia chetosi, acidosi metabolica ipoglicemia rifiuto dell’alimentazione o difficoltà nell’allattamento ipotonia convulsioni bassa temperatura corporea tachipnea ..E quindi procedere al dosaggio degli indici di flogosi per escludere una sepsi, quindi dell’ammoniemia, delle transaminasi e degli aminoacidi plasmatici ed urinari. Saudubray JM et al. Clinical approach to Inherited Metabolic Diseases in the neonatal period: A 20 year survey. J Inherit Metab Dis 1989; 12 (s1): ) Neonato: uMol/L Lattante-Bambino: uMol/L Donna: uMol/L Uomo: uMol/L Valori normali di AMMONIEMIA Haberle (2011) Eur J Pediatr 170:221-34
: ) Neonato: uMol/L. Lattante-Bambino: uMol/L. Donna: uMol/L. Uomo: uMol/L. Valori normali di. AMMONIEMIA. Haberle (2011) Eur J Pediatr 170:")
16
Dato che l’Ammonio attraversa facilmente e rapidamente la BEE…

17
Encefalopatia da iperammoniemia
Aumento flusso cerebrale Edema da glutamina Alterazione della BEE Iperpolarizzazione delle membrane Alterazione dei neurotrasmettitori Gliosi con astrociti Lesioni della sostanza bianca (alterata mielinizzazione e lesioni cistiche) Atrofia corticale Lesioni spongiformi cortico-sottocorticali, talamiche e dei nuclei della base J Inherit Metab Dis :
 Atrofia corticale. Lesioni spongiformi cortico-sottocorticali, talamiche e dei nuclei della base. J Inherit Metab Dis :")
18
Diagnosi prenatale Tutti e cinque gli errori congeniti dell'ureagenesi possono essere diagnosticati prima della nascita: dosaggio di metaboliti anomali nel liquido amniotico analisi del DNA dai villi coriali o dagli amniociti valutazione dell'attività enzimatica o biopsia epatica in utero. (C. Scriver et al., The Metabolic and Molecular Bases of Inherited Disease, Eighth Edition). Prevenzione dell'iperammoniemia neonatale nei neonati a rischio o con parente affetto: Protocollo diagnostico-terapeutico che mira a prevenire l'iperammoniemia dosando citrullina e argininsuccinato a 60 ore di vita. Nonostante la prognosi di questi bambini sia sicuramente migliore rispetto a quella dei bambini trattati dopo che l'iperammoniemia si sia verificata, i risultati sono deludenti in quanto molti presentano qualche difficoltà di sviluppo.
. Prevenzione dell iperammoniemia neonatale nei neonati a rischio o con parente affetto: Protocollo diagnostico-terapeutico che mira a prevenire l iperammoniemia dosando citrullina e argininsuccinato a 60 ore di vita. Nonostante la prognosi di questi bambini sia sicuramente migliore rispetto a quella dei bambini trattati dopo che l iperammoniemia si sia verificata, i risultati sono deludenti in quanto molti presentano qualche difficoltà di sviluppo.")
19
Obiettivo della terapia: evitare l’iperammoniemia elevata persistente, in quanto..
Il riconoscimento precoce e il tempestivo inizio del trattamento consentono di prevenire il danno neuronale irreversibile…

20
…e di evitare la morte del bambino
Outcome neurologico dei difetti del ciclo dell’urea -studio giapponese- Uchino t et al. J Inherit Metab. Dis. 1998; 21 Suppl. 1:

21
Lo scopo della terapia dei difetti del ciclo dell'urea:
Fornire una dieta con apporto sufficiente di proteine, arginina, ed energia per la crescita e lo sviluppo ed allo stesso tempo prevenire i disturbi metabolici, iperammoniemia e iperglutaminemia. Nella fase acuta Emodialisi/emofiltrazione, dialisi peritoneale elevato apporto calorico con basso apporto di proteine (0,7 mg/kg/die) utilizzo di sostanze che favoriscono la detossicazione dell’ammonio (sodio benzoato rimuove la glicina; sodio fenilbutirrato) Fenilacetato di sodio Arginina (attiva il NAGS) Acido Carglumico (attiva CPS1) In cronico: Dieta con basso apporto di proteine integrazione con aminoacidi essenziali sodio benzoato e fenilbutirrato
 utilizzo di sostanze che favoriscono la detossicazione dell’ammonio (sodio benzoato rimuove la glicina; sodio fenilbutirrato) Fenilacetato di sodio. Arginina (attiva il NAGS) Acido Carglumico (attiva CPS1) In cronico: Dieta con basso apporto di proteine. integrazione con aminoacidi essenziali. sodio benzoato e fenilbutirrato.")
22
Per concludere… IN UN NEONATO CHE “STA MALE” O “CHE E’ STRANO”:
anamnesi familiare eseguire in prima istanza EGA e ammoniemia IN UN NEONATO CON IPERAMMONIEMIA (>100mmol/L): sospensione immediata del latte glucosata e.v. NEONATO CON IPERAMMONIEMIA CONFERMATA: invio verso centro specializzato per malattie metaboliche inizio immediato arginina e sodio benzoato farmaci “speciali” / emodialisi ?
: sospensione immediata del latte. glucosata e.v. NEONATO CON IPERAMMONIEMIA CONFERMATA: invio verso centro specializzato per malattie metaboliche. inizio immediato arginina e sodio benzoato. farmaci speciali / emodialisi")
23
Grazie...

Presentazioni simili

>")


















