Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
EMOCOAGULAZIONE

2
EMOSTASI Serie di reazioni biochimiche e cellulari, sequenziali e sinergiche, finalizzate a impedire la perdita di sangue dai vasi. E’ cioè un meccanismo di difesa deputato al mantenimento dell’integrità dei vasi sanguigni e della fluidità del sangue. Alterazioni dell’emostasi AUMENTO RIDUZIONE TROMBOSI EMORRAGIA

3
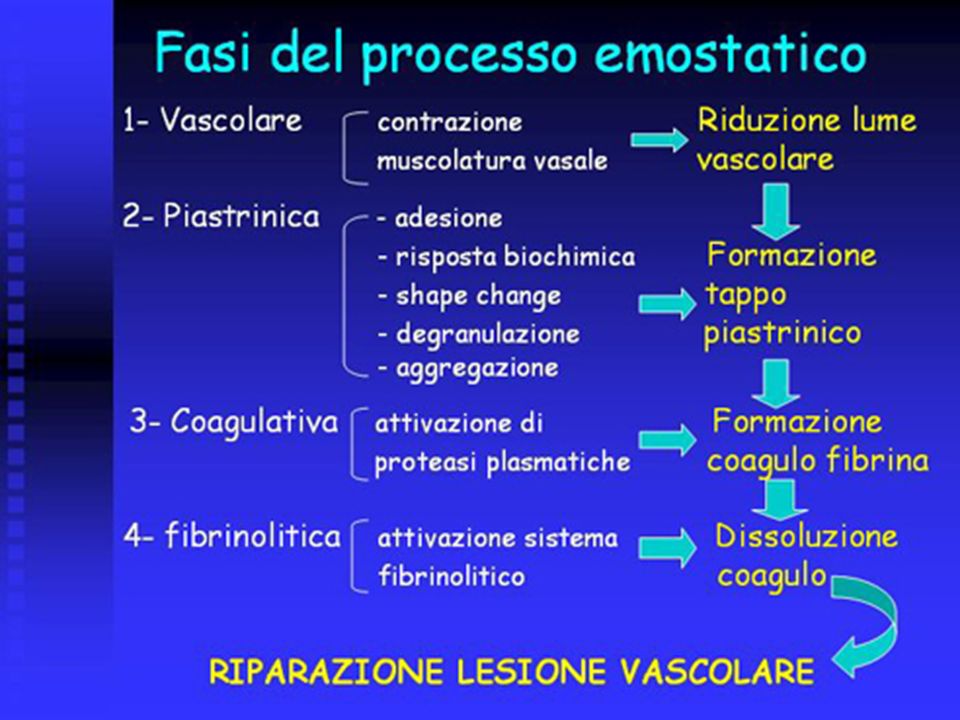
Tappe del processo emostatico
Quando si verifica una lesione vascolare, viene attivato il meccanismo dell’emostasi, che è un meccanismo autoregolato. I sistemi coinvolti nel processo emostatico sono 4: Vasi e costituenti della parete vascolare Piastrine Cascata enzimatica della coagulazione Sistema fibinolitico

5
FASE VASCOLARE CONTRAZIONE VASALE
Contrazione delle cellule muscolari lisce dei vasi Riflesso neurovegetativo vasomotore (nerva vasorum) SOSTANZE VASOCOSTRITTRICI Endotelina Serotonina ed istamina
 SOSTANZE VASOCOSTRITTRICI. Endotelina. Serotonina ed istamina.")
6
FASE PIASTRINICA FORMAZIONE DEL TAPPO PIASTRINICO (si forma in 3-5 minuti)
L’endotelio è un tessuto metabolicamente attivo, che, a seconda del suo stato funzionale, può o favorire o inibire l’emostasi. In stato di quiescenza l’endotelio è in grado di assicurare la fluidità del sangue mediante un complesso meccanismo anticoagulante mentre, in seguito ad una lesione, la perdita della cellula endoteliale costituisce il punto di avvio del processo di emostasi localizzata, attraverso l’induzione coordinata di attività pro-emostatiche che iniziano con l’adesione piastrinica.

7
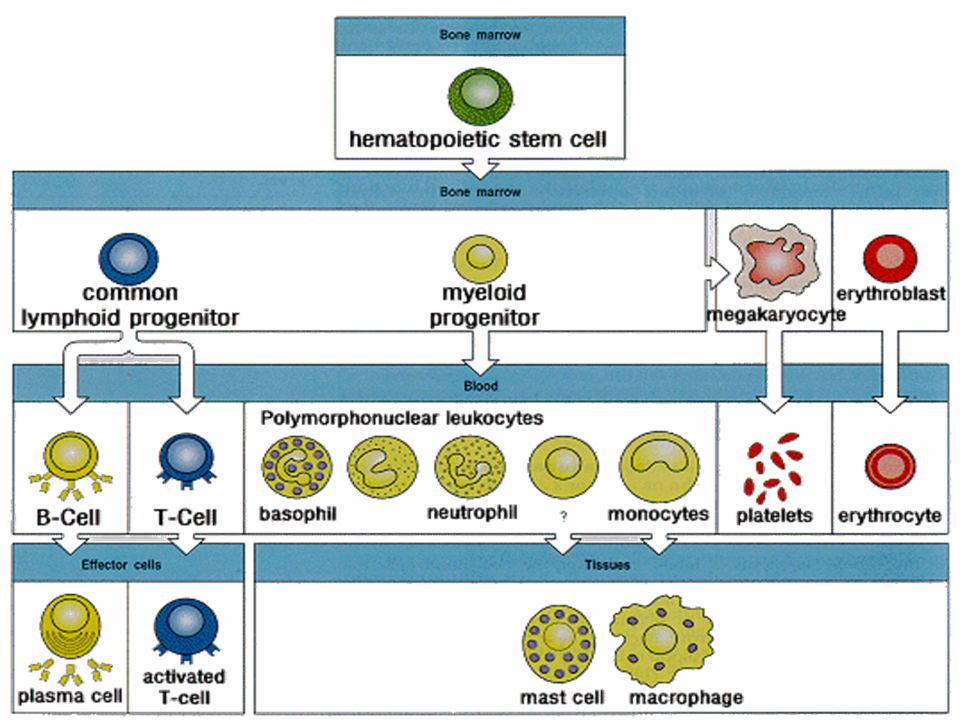
PIASTRINE normali: 150,000 - 400,000 per ul
Sito d’accumulo nella milza (30%) vita giorni Fagocitati dai neutrofi e dai monociti/macrofagi Derivano dai megacariociti 30,000 al giorno
 vita giorni. Fagocitati dai neutrofi e dai monociti/macrofagi. Derivano dai megacariociti. 30,000 al giorno.")
9
Le piastrine sono frammenti citoplasmatici di una cellula progenitrice midollare multinucleata, il megacariocita. La trombopoiesi o piastrinopoiesi è regolata da un fattore presente nel siero, la trombopoietina, che è in grado di aumentare non solo la produzione di piastrine, ma anche la proliferazione dei megacariociti. Anche l’interleuchina-11 (IL-11) ha attività trombopoietica. La produzione di piastrine può aumentare notevolmente (7-8 volte) in seguito ad attivazione dell’emostasi o a stimolazione del midollo. Le piastrine appena immesse in circolo sono più grandi ed hanno un’attività emostatica maggiore rispetto alle piastrine circolanti mature. Le piastrine sopravvivono in circolo per circa giorni (emivita 5-6 giorni) e successivamente vengono sequestrate dagli organi emocateretici (principalmente dalla milza e dal fegato), dove vengono fagocitate dalle cellule del sistema dei fagociti mononucleati.
 ha attività trombopoietica. La produzione di piastrine può aumentare notevolmente (7-8 volte) in seguito ad attivazione dell’emostasi o a stimolazione del midollo. Le piastrine appena immesse in circolo sono più grandi ed hanno un’attività emostatica maggiore rispetto alle piastrine circolanti mature. Le piastrine sopravvivono in circolo per circa giorni (emivita 5-6 giorni) e successivamente vengono sequestrate dagli organi emocateretici (principalmente dalla milza e dal fegato), dove vengono fagocitate dalle cellule del sistema dei fagociti mononucleati.")
10
La loro forma è controllata dal citoscheletro e in particolare da un fascio circonferenziale di microtubuli, situato all’equatore del disco, e da microfilamenti contrattili ancorati alle membrane cellulari. A parte l’assenza del nucleo, sono presenti tutti i principali componenti subcellulari, mitocondri, granuli di glicogeno, lisosomi. La membrana plasmatica è rivestita all’esterno da un caratteristico strato di polisaccaridi e lipo/glicoproteine, detto glicocalice. Del glicocalice fanno parte i recettori, che mediano le più importanti funzioni piastriniche e tutte le glicoproteine coinvolte nell’adesione e nell’aggregazione. Le piastrine contengono tre tipi di granuli:lisosomi, granuli densi (detti anche granuli delta) ed alfa-granuli. Il contenuto di questi ultimi due viene secreto durante la risposta piastrinica, attraverso il sistema canalicolare aperto o attraverso la membrana plasmatica, direttamente nel microambiente sede dell’aggregazionepiastrinica
 ed alfa-granuli. Il contenuto di questi ultimi due viene secreto durante la risposta piastrinica, attraverso il sistema canalicolare aperto o attraverso la membrana plasmatica, direttamente nel microambiente sede dell’aggregazionepiastrinica.")
11
PIASTRINA

12
Fasi della risposta piastrinica
In seguito al danno vascolare, le piastrine sono esposte al sottoendotelio, cioè collageno, proteoglicani, fibronectina ed altre glicoproteine e questo ne determina l’attivazione. La risposta piastrinica comporta mutamenti di ordine biochimico, strutturale e morfologico delle piastrine stesse. La risposta delle piastrine ad uno stimolo è dovuta all’intervento coordinato della membrana, dei granuli e del citoscheletro e può essere suddivisa in varie fasi che tendono a sovrapporsi.

14
Adesione L’endotelio integro e la superficie piastrinica si respingono in virtù delle loro cariche negative La rottura dell’endotelio espone il collageno sottoendoteliale che lega la GpIa. La GpIb si lega al vWF, a sua volta adeso al collageno

15
Cambiamento di forma In seguito all’adesione le piastrine attivano meccanismi di trasduzione che determinano il cambiamento di forma e la reazione di degranulazione. Fase reversibile Refrattarietà piastrinica Fase irreversibile

16
Cambiamento di forma Rende disponibile il fattore piastrinico 3 (F.P.3) una fosfatidil-serina che nella piastrina a riposo è situata sul versante interno della membrana piastrinica che ha attività procoagulante. I fosfolipidi della membrana piastrinica forniscono la fase solida sulla quale avvengono le interazioni fra I vari fattori della coagulazione, quando la cascata coagulativa viene attivata.
 una fosfatidil-serina che nella piastrina a riposo è situata sul versante interno della membrana piastrinica che ha attività procoagulante. I fosfolipidi della membrana piastrinica forniscono la fase solida sulla quale avvengono le interazioni fra I vari fattori della coagulazione, quando la cascata coagulativa viene attivata.")
17
Release piastrinico È dipendente dall’ATP e dal citoscheletro
La secrezione delle piastrine avviene subito dopo l’adesione ed è un fenomeno attivo (legato anche all’aumento della concentrazione di calcio nelle cellule) che determina il rilascio del contenuto dei granuli piastrinici all’esterno. È dipendente dall’ATP e dal citoscheletro
 che determina il rilascio del contenuto dei granuli piastrinici all’esterno. È dipendente dall’ATP e dal citoscheletro.")
19
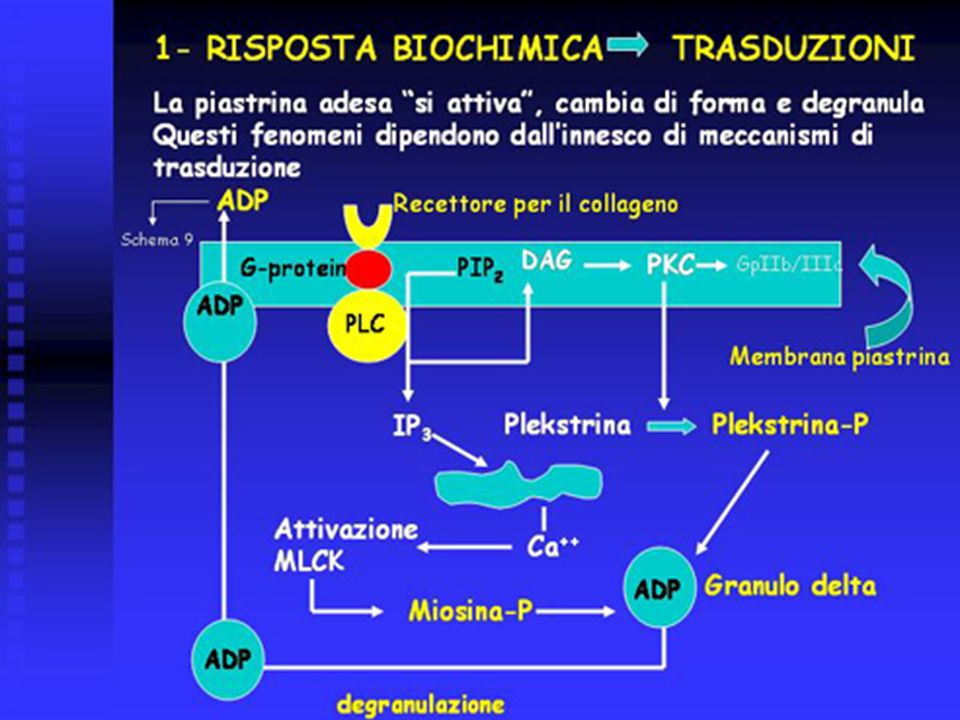
L’adesione piastrinica al collageno avviene tramite recettori specifici che sono accoppiati a proteine G di trasduzione. Tali proteine attivano la fosfolipasi C (PLC), la quale idrolizza il fosfatidil - inositolo di membrana (PIP2) a inositolo – trifosfato (IP3) e diacil – glicerolo (DAG). L’IP3 libera il Ca dai depositi non mitocondriali ed il Ca attiva la cinasi della catena leggera della miosina (MLCK), con fosforilazione della miosina (miosina - P). Il DAG attiva la proteino – cinasi C (PKC), la quale fosforila la plekstrina (plekstrina - P). Plekstrina – P e miosina – P inducono degranulazione piastrinica con rilascio di ADP dai granuli delta.
, con fosforilazione della miosina (miosina - P). Il DAG attiva la proteino – cinasi C (PKC), la quale fosforila la plekstrina (plekstrina - P). Plekstrina – P e miosina – P inducono degranulazione piastrinica con rilascio di ADP dai granuli delta.")
21
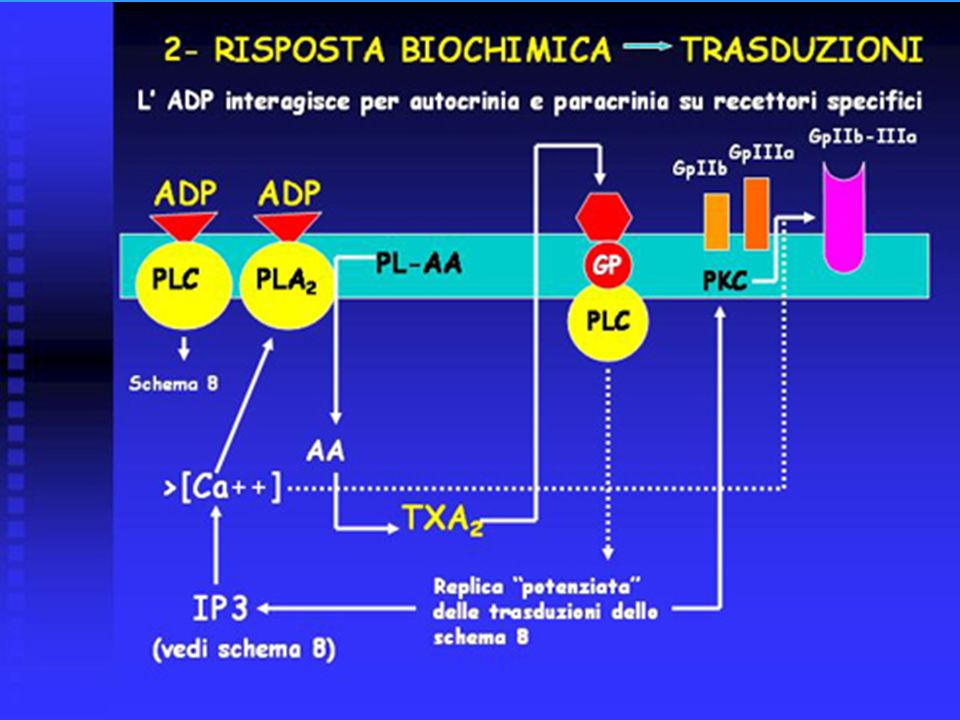
L’ADP liberato si lega a due tipi di recettori specifici, accoppiati con PLC e con PLA2.
Anche il Ca liberato attiva la fosfolipasi A2 (PL2), che è anche accoppiata allo stesso recettore per ADP. Tale fosfolipasi stacca l’acido arachidonico (AA) dai fosfolipidi di membrana (PL - AA). L’AA viene elaborato dalla trombossano sintetasi delle piastrine e trasformato in trombossano A2 (TXA2). Una potente ondata di TXA2 viene rilasciata dalle piastrine ed interagisce con recettori specifici (autocrinia, paracrinia), accoppiati con una proteina G che attiva la PLC. Si replica quindi, con maggiore intensità, la via di trasduzione innescata dalla interazione con il collageno. Contemporaneamente, il Ca liberato da IP3 promuove assieme alla PKC, la fusione delle proteine GpIIb e GpIIIa a formare il complesso glicoproteico GpIIb-IIIa, recettore per il fibrinogeno. Il fibrinogeno si pone a ponte fra le piastrine ed ha quindi avvio la fase di aggregazione piastrinica.
, che è anche accoppiata allo stesso recettore per ADP. Tale fosfolipasi stacca l’acido arachidonico (AA) dai fosfolipidi di membrana (PL - AA). L’AA viene elaborato dalla trombossano sintetasi delle piastrine e trasformato in trombossano A2 (TXA2). Una potente ondata di TXA2 viene rilasciata dalle piastrine ed interagisce con recettori specifici (autocrinia, paracrinia), accoppiati con una proteina G che attiva la PLC. Si replica quindi, con maggiore intensità, la via di trasduzione innescata dalla interazione con il collageno. Contemporaneamente, il Ca liberato da IP3 promuove assieme alla PKC, la fusione delle proteine GpIIb e GpIIIa a formare il complesso glicoproteico GpIIb-IIIa, recettore per il fibrinogeno. Il fibrinogeno si pone a ponte fra le piastrine ed ha quindi avvio la fase di aggregazione piastrinica.")
23
L’aggregazione primaria (prima onda di aggregazione), è un’aggregazione reversibile, indotta da piccole quantità di agonisti che interagiscono con i loro recettori sulla membrana piastrinica (ADP, collageno, trombina, PAF, ecc.)
, è un’aggregazione reversibile, indotta da piccole quantità di agonisti che interagiscono con i loro recettori sulla membrana piastrinica (ADP, collageno, trombina, PAF, ecc.)")
24
L’aggregazione secondaria (seconda onda di aggregazione) è dovuta invece sia all’interazione di grosse quantità di agonisti con i loro recettori, sia al rilascio di grosse quantità di ADP e quindi di TXA2 da parte delle piastrine attivate da piccole quantità di agonisti molto potenti. Si forma dapprima un aggregato piastrinico che prende il nome di TAPPO EMOSTATICO TEMPORANEO O PRIMARIO, che è reversibile. Successivamente, dove vi è stata adesione e aggregazione primaria si ha la formazione di un aggregato impermeabile e irreversibile, detto TAPPO EMOSTATICO SECONDARIO.

25
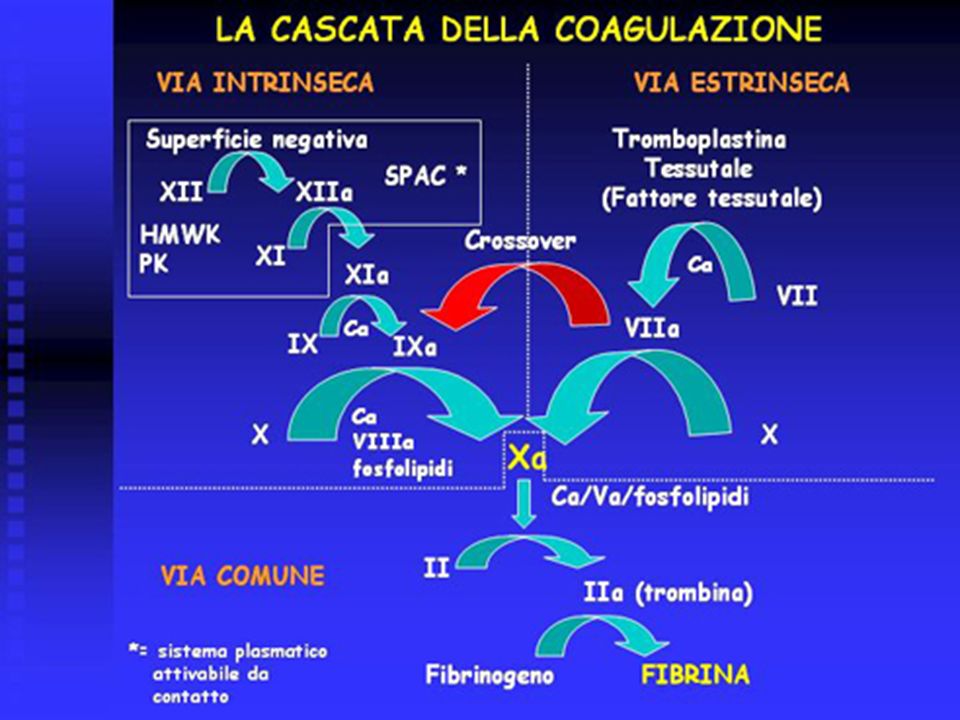
Coagulazione E’ il terzo componente del processo emostatico e porta alla formazione del coagulo insolubile di fibrina, derivante dalla trasformazione del precursore plasmatico solubile fibrinogeno. A questo risultato si giunge grazie all’attivazione sequenziale di una serie di fattori plasmatici (fattori della coagulazione). I fattori plasmatici, ad eccezione della pre-callicreina (PK) e del chininogeno ad alto peso molecolare (high molecular weight kininogen, HMWK), sono numerati progressivamente dall’1 al 13, secondo una nomenclatura internazionale . Tutti i fattori della coagulazione, ad eccezione del fattore III (fattore tessutale), sono normalmente presenti nel plasma in forma inattiva.
. I fattori plasmatici, ad eccezione della pre-callicreina (PK) e del chininogeno ad alto peso molecolare (high molecular weight kininogen, HMWK), sono numerati progressivamente dall’1 al 13, secondo una nomenclatura internazionale . Tutti i fattori della coagulazione, ad eccezione del fattore III (fattore tessutale), sono normalmente presenti nel plasma in forma inattiva.")
28
Fattori della coagulazione

30
Sistema fibrinolitico
La formazione della fibrina si verifica nel corso di vari processi, come l’infiammazione, la riparazione delle ferite e, soprattutto, l’emostasi e deve essere limitata nello spazio e nel tempo una volta che lo stimolo scatenante abbia terminato di agire. La fibrinolisi rappresenta il meccanismo fondamentale attraverso il quale si dissolve il coagulo di fibrina, dopo che ha svolto la sua funzione.

31
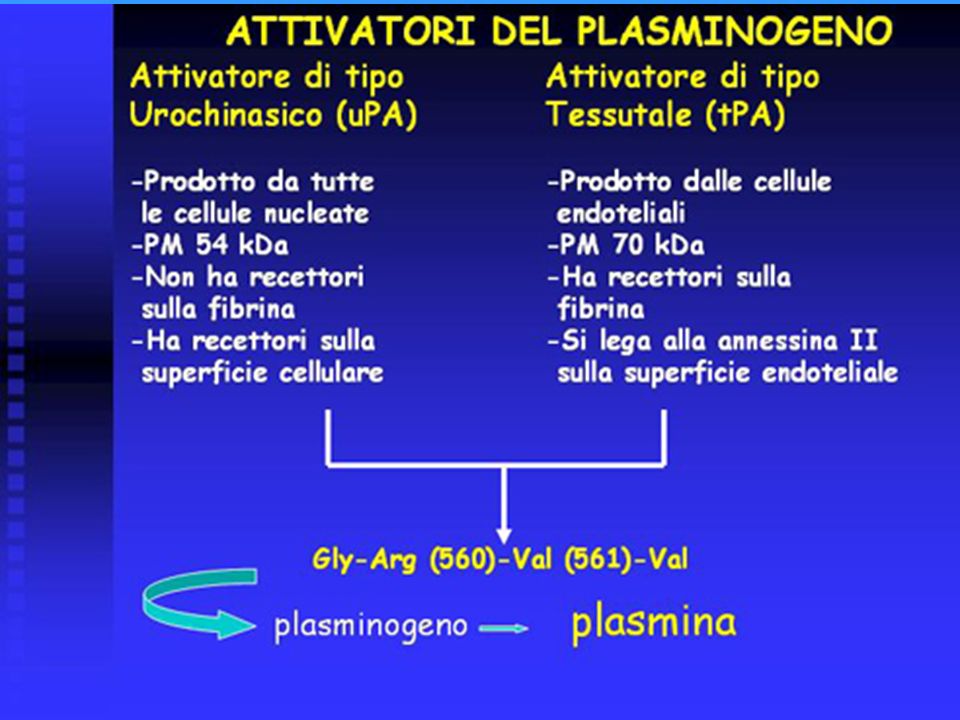
Il sistema fibrinolitico è un SISTEMA MULTIENZIMATICO, che presenta analogie con il sistema della coagulazione. E’ infatti costituito da serino-proteasi (sito attivo composto da serina-acido aspartico- istidina). Il sito catalitico si trova nella regione C-terminale (catena B), mentre la regione N-terminale (catena A) contiene uno o più domini funzionali, responsabili delle diverse funzioni di queste molecole, come ad es. legame alla fibrina, legame a recettori sulle superfici cellulari, legame al plasminogeno, ecc. Sono enzimi tripsino-simili, cioè agiscono a livello del legame specifico arginina-lisina. Si trovano in forma di zimogeni, che vengono trasformati in enzimi attivi mediante un taglio proteolitico. La “reazione centrale” della fibrinolisi è rappresentata dalla conversione del plasminogeno (pro-enzima plasmatico, inattivo) nell’enzima proteolitico attivo plasmina, mediante la scissione di un singolo legame peptidico. La plasmina così prodotta degrada la fibrina, dando origine a prodotti di degradazione solubili e quindi alla lisi del coagulo di fibrina. Componenti del sistema fibrinolitico: - attivatori del plasminogeno (attivatore tissutale o tPA; attivatore di tipo urochinasico o uPA) - plasminogeno - plasmina - inibitori
 nell’enzima proteolitico attivo plasmina, mediante la scissione di un singolo legame peptidico. La plasmina così prodotta degrada la fibrina, dando origine a prodotti di degradazione solubili e quindi alla lisi del coagulo di fibrina. Componenti del sistema fibrinolitico: - attivatori del plasminogeno. (attivatore tissutale o tPA; attivatore di tipo urochinasico o uPA) - plasminogeno. - plasmina. - inibitori.")
35
Fibrinolisi Plasminogeno convertito a plasmina
Plasmina degrada la fibrina Rilascio di fattori della degradazione Plasmina è inattivata da inibitori in circolazione

37
Patologie della coagulazione
CAUSE deficit della parete vasale disfunzione o deficienza delle piastrine deregolamentazione o deficit di fattori della coagulazione

38
Difetti delle piastrine

39
Trombofilie ereditarie
La trombofilia identifica una tendenza a sviluppare trombosi conseguenti ad alterazioni del sistema coagulativo o fibrinolitico su base ereditaria o acquisita Riconoscono una modalità di trasmissione genetica di tipo autosomico dominante Si caratterizzano per episodi di trombosi prevalentemente venosi Assenza di fattori di rischio Esordio della trombosi in età giovanile (al di sotto di 40-50aa) Gli episodi di trombosi tendono a recidivare E’ spesso presente una storia familiare Aborti ripetuti e/o feti nati morti
 Gli episodi di trombosi tendono a recidivare. E’ spesso presente una storia familiare. Aborti ripetuti e/o feti nati morti.")
40
Fattori di rischio acquisiti
DEFICIT DEGLI INIBITORI NATURALI DELLA COAGULAZIONE AT, PC, PS <1% della popolazione e <10% di soggetti non selezionati con TVP Rischio di TVP è 5-8% Incidenza annua 1-2% Circa il 50% degli episodi di TVP concomita con eventi occasionali Frequente esordio <45aa

41
Presente solo negli individui caucasici
Fattore V di Leiden(G1691A) (arginina 506 è sostituita con ac.glutammico-cromosoma 1) Presente solo negli individui caucasici Prevalenza del 2-15% (40% in pts con trombosi) Rischio di TVP 5-8% negli eterozigoti, negli omozigoti Incidenza annua di TVP 0.19%-0.67% Gravidanza, contraccettivi orali, chirurgia minore, viaggi prolungati, malattie intercorrenti Frequente esordio dopo i 45 aa PROTROMBINA G20120A mutazione nella regione 3’ non tradotta I portatori hanno una concentrazione plasmatica della protrombina superiore del 30%(trascrizione +efficiente e/o >stabilità del messaggio) Prevalenza del 2% negli individui sani e 20% dei pazienti con TVP Rischio del 2-5% negli eterozigoti Il rischio di TVP aumenta con l’età fino ad un massimo del 19% dopo 60 aa
 (arginina 506 è sostituita con ac.glutammico-cromosoma 1) Presente solo negli individui caucasici. Prevalenza del 2-15% (40% in pts con trombosi) Rischio di TVP 5-8% negli eterozigoti, negli omozigoti. Incidenza annua di TVP 0.19%-0.67% Gravidanza, contraccettivi orali, chirurgia minore, viaggi prolungati, malattie intercorrenti. Frequente esordio dopo i 45 aa. PROTROMBINA G20120A mutazione nella regione 3’ non tradotta I portatori hanno una concentrazione plasmatica della protrombina superiore del 30%(trascrizione +efficiente e/o >stabilità del messaggio) Prevalenza del 2% negli individui sani e 20% dei pazienti con TVP. Rischio del 2-5% negli eterozigoti. Il rischio di TVP aumenta con l’età fino ad un massimo del 19% dopo 60 aa.")
42
È un fattore di rischio per trombosi sia arteriose che venose
IPEROMOCISTEINEMIA (è un sulfidril aa derivato dalla conversione metabolica della metionina) È un fattore di rischio per trombosi sia arteriose che venose Si associa al polimorfismo C677T del gene della metilentetraidrofolato reduttasi (omozigote). Fattori acquisiti (ridotta assunzione di piridossina, cobalamina, folati) si sommano al difetto genetico La prevalenza del gene TT nella popolazione generale e nei pazienti con TVP è del 13% ALTRI FATTORI DI RISCHIO I difetti combinati (fattoreV+G20120A) duplicano il rischio di TVP (20 volte) L’iperomocisteinemia lieve+ fattore V o G20120A aumentano il rischio di 20/50 volte Aplotipo HR2 omozigote del fattore V costituisce una causa di resistenza della proteina C attivata (rischio del 5%) Aumentati livelli di fibrinogeno fattore VIII fattore IX e fattore XI, inibitore della fibrinolisi trombino attivabile (TAF1)
 È un fattore di rischio per trombosi sia arteriose che venose. Si associa al polimorfismo C677T del gene della metilentetraidrofolato reduttasi (omozigote). Fattori acquisiti (ridotta assunzione di piridossina, cobalamina, folati) si sommano al difetto genetico. La prevalenza del gene TT nella popolazione generale e nei pazienti con TVP è del 13% ALTRI FATTORI DI RISCHIO I difetti combinati (fattoreV+G20120A) duplicano il rischio di TVP (20 volte) L’iperomocisteinemia lieve+ fattore V o G20120A aumentano il rischio di 20/50 volte. Aplotipo HR2 omozigote del fattore V costituisce una causa di resistenza della proteina C attivata (rischio del 5%) Aumentati livelli di fibrinogeno fattore VIII fattore IX e fattore XI, inibitore della fibrinolisi trombino attivabile (TAF1)")
43
MANIFESTAZIONI CLINICHE
TVP degli arti inferiori con o senza embolia polmonare (circa 40% dei casi) Trombosi venose superficiali, del circolo splancnico e cerebrali sono più rare Le trombofilie ereditare sono responsabili di circa il 40% delle TVP non provocate da fattori acquisiti Le trombosi arteriose possono associarsi a protrombina G20120A nei soggetti giovani e all’iperomocisteinemia RECIDIVA DI TVP I rischio di recidiva è aumentato nei soggetti con trombofilia ereditaria anche a distanza di anni dal primo episodio La recidiva ha prognosi infausta nel 5% dei pazienti Il 30% dei pazienti cin recidiva sviluppa una sindrome post-trombotica Il rischio di recidiva è maggiore nei soggetti con difetto degli anticoagulaanti naturali, nelle doppie eterozigosi e nella omozigosi per il fattore V di Leiden
 Trombosi venose superficiali, del circolo splancnico e cerebrali sono più rare Le trombofilie ereditare sono responsabili di circa il 40% delle TVP non provocate da fattori acquisiti Le trombosi arteriose possono associarsi a protrombina G20120A nei soggetti giovani e all’iperomocisteinemia. RECIDIVA DI TVP I rischio di recidiva è aumentato nei soggetti con trombofilia ereditaria anche a distanza di anni dal primo episodio La recidiva ha prognosi infausta nel 5% dei pazienti Il 30% dei pazienti cin recidiva sviluppa una sindrome post-trombotica Il rischio di recidiva è maggiore nei soggetti con difetto degli anticoagulaanti naturali, nelle doppie eterozigosi e nella omozigosi per il fattore V di Leiden")
44
SCREENING FAMILIARE Elevata probabilità di identificare portatori (50%) Profilassi dei portatori in condizioni di rischio Identificazione precoce di eventi spontanei Etichetta di malattia genetica Possibili ripercussioni assicurative Discriminazioni lavorative Bassa incidenza di TVP

45
SCREENING di primo livello
Antitrombin heparin cofactor activity (amidolytic method) Protein C (clotting or amidolytic method) Protein s (total and free antigen fractions) Factor V Leiden Prothrombin G20120A Homocysteine
 Protein C (clotting or amidolytic method) Protein s (total and free antigen fractions) Factor V Leiden. Prothrombin G20120A. Homocysteine.")
46
TERAPIA Non differisce dai pazienti con TVP senza trombofilia ereditaria Il deficit di AT richiede elevate dosi di eparina per raggiungere livelli terapeutici di aPTT (considerare l’utilizzo di concentrati di AP purificata ) Un severo difetto omozigote di PC necessita di un aggiunta di concentrati di PC nella fase di transizione fra la terapia con eparina e quella con anticoagulanti orali per mantenere livelli di PC sopra il 50% fino al raggiungimento di una anticoagulazione stabile (rischio necrosi cutanea)
 Un severo difetto omozigote di PC necessita di un aggiunta di concentrati di PC nella fase di transizione fra la terapia con eparina e quella con anticoagulanti orali per mantenere livelli di PC sopra il 50% fino al raggiungimento di una anticoagulazione stabile (rischio necrosi cutanea)")
47
ESAMI DI LABORATORIO

48
TEST DI SCREENING misurano gli effetti combinati di fattori che influenzano una fase particolare della coagulazione (p. es., il tempo di sanguinamento). Dosaggi specifici misurano il livello o la funzione di un fattore dell'emostasi (p. es., il dosaggio del fattore VIII). Test addizionali che possono misurare un prodotto o un effetto di un'attivazione patologica in vivo delle piastrine, della coagulazione o della fibrinolisi (p. es., il livello dei prodotti di degradazione della fibrina). I risultati dei test di screening e la conoscenza del disordine clinico fanno da guida nella selezione di indagini diagnostiche più specifiche. Il tempo di sanguinamento deve essere determinato con un manicotto dello sfigmomanometro applicato all'arto superiore gonfiato fino a 40 mm Hg, per far sì che il coagulo emostatico venga tenuto contro una retropressione. Un dispositivo a molla monouso per il rilievo del tempo di sanguinamento viene utilizzato per fare un'incisione di 6-mm× 1 mm sulla parte volare dell'avambraccio. Si assorbe il sangue con l'orlo di una carta da filtro a intervalli di 30 s finché il sanguinamento non si arresti. Con questo metodo il limite superiore della norma per il tempo di sanguinamento è di 7,5 min. La trombocitopenia, disordini della funzione piastrinica e la malattia di von Willebrand (VWD) prolungano il tempo di sanguinamento, il quale invece non risulta prolungato nei disordini della fase della coagulazione. L'uso dell'aspirina nei 5-7 giorni precedenti prolunga anche il tempo di sanguinamento.
. Dosaggi specifici misurano il livello o la funzione di un fattore dell emostasi (p. es., il dosaggio del fattore VIII). Test addizionali che possono misurare un prodotto o un effetto di un attivazione patologica in vivo delle piastrine, della coagulazione o della fibrinolisi (p. es., il livello dei prodotti di degradazione della fibrina). I risultati dei test di screening e la conoscenza del disordine clinico fanno da guida nella selezione di indagini diagnostiche più specifiche. Il tempo di sanguinamento deve essere determinato con un manicotto dello sfigmomanometro applicato all arto superiore gonfiato fino a 40 mm Hg, per far sì che il coagulo emostatico venga tenuto contro una retropressione. Un dispositivo a molla monouso per il rilievo del tempo di sanguinamento viene utilizzato per fare un incisione di 6-mm× 1 mm sulla parte volare dell avambraccio. Si assorbe il sangue con l orlo di una carta da filtro a intervalli di 30 s finché il sanguinamento non si arresti. Con questo metodo il limite superiore della norma per il tempo di sanguinamento è di 7,5 min. La trombocitopenia, disordini della funzione piastrinica e la malattia di von Willebrand (VWD) prolungano il tempo di sanguinamento, il quale invece non risulta prolungato nei disordini della fase della coagulazione. L uso dell aspirina nei 5-7 giorni precedenti prolunga anche il tempo di sanguinamento.")
49
Il tempo di tromboplastina parziale (PTT) individua i casi di reazioni emocoagulative anomale innescate dall'esposizione del plasma a una superficie carica negativamente. Il plasma viene incubato per 3 min con un reagente che fornisce il fosfolipide procoagulante e una polvere tensioattiva (p. es., silicio micronizzato). Si aggiunge quindi Ca e si valuta il tempo di coagulazione. (Dal momento che i reagenti in commercio e la strumentazione variano notevolmente, ciascun laboratorio deve determinare il proprio range di normalità; di regola da 28 a 34 sec). Il PTT è sensibile a deficit di circa il 30-40% di tutti i fattori della coagulazione eccetto che per i fattori VII e XIII. Con rare eccezioni, un risultato normale esclude l'emofilia. L'eparina prolunga il PTT e quest'ultimo viene utilizzato spesso per il monitoraggio della terapia eparinica. Un tempo prolungato può anche essere causato da un deficit di uno o più fattori della coagulazione o dalla presenza di un inibitore di un fattore della coagulazione (p. es., un anticoagulante che inibisce il fattore VIII, v. Disordini della coagulazione da anticoagulanti circolanti, più avanti) o di un inibitore del fosfolipide procoagulante (inibitore del lupus-v. Disordini della coagulazione da anticoagulanti circolanti, oltre). Se è presente un inibitore, mescolando il plasma del paziente 1:1 con un plasma normale, non risulterà un accorciamento del PTT entro i 5 s circa del tempo che si ottiene con il plasma normale da solo. Determinazioni degli specifici fattori della coagulazione possono in genere individuare la causa di un prolungato PTT, non prontamente spiegato sulla base degli altri rilievi clinici.

50
Nel test del tempo di protrombina (PT), il plasma viene ricalcificato in presenza di un'alta concentrazione di fattore tissutale (tromboplastina tissutale). Il test individua i casi in cui siano presenti anomalie dei fattori V, VII e X, della protrombina e del fibrinogeno; il PT normale varia da 10 e 12 sec, in rapporto al particolare reagente impiegato contenente fattore tissutale e di altri dettagli tecnici. Un PT più lungo di 2 s rispetto al valore normale, di controllo del laboratorio, deve essere considerato anormale e ne va indagato il motivo. Il PT è un esame utile per lo screening di disturbi della coagulazione in varie condizioni acquisite (p. es., carenza di vitamina K, epatopatie, CID). Il PT viene anche impiegato per controllare la terapia con anticoagulanti cumarinici. Il range terapeutico del PT dipende dalla tromboplastina utilizzata in ogni laboratorio. APTT :tempo di protrombina parziale attivato
. Il PT viene anche impiegato per controllare la terapia con anticoagulanti cumarinici. Il range terapeutico del PT dipende dalla tromboplastina utilizzata in ogni laboratorio. APTT :tempo di protrombina parziale attivato.")
51
Per determinare il tempo di trombina, il plasma da saggiare e un plasma normale di controllo vengono fatti coagulare con l'aggiunta di un reagente contenente trombina bovina diluito in modo tale da dare un tempo di coagulazione di circa 15 sec per il plasma di controllo. Poiché il test è indipendente dalle reazioni che generano trombina, esso è usato per riconoscere specificamente le anomalie che interessano la reazione trombina-fibrinogeno: eparina, presenza in alta quantità di prodotti di degradazione della fibrina e alterazioni qualitative del fibrinogeno. È particolarmente utile per accertare se un campione di plasma contenga eparina (p. es., eparina residua non neutralizzata dopo un intervento di bypass extracorporeo o sangue contaminato prelevato tramite dispositivi tenuti pervi mediante lavaggi con eparina). Nel plasma che contiene eparina, il tempo di trombina risulta prolungato, ma quando il test viene ripetuto, sostituendo la trombina con la batroxobina (un enzima del veleno di serpente insensibile all'eparina e che converte direttamente il fibrinogeno in fibrina), il test risulterà normale.
. Nel plasma che contiene eparina, il tempo di trombina risulta prolungato, ma quando il test viene ripetuto, sostituendo la trombina con la batroxobina (un enzima del veleno di serpente insensibile all eparina e che converte direttamente il fibrinogeno in fibrina), il test risulterà normale..")
52
La stabilità del coagulo di fibrina viene saggiata facendo coagulare 0,2 ml di plasma con 0,2 ml di cloruro di Ca e incubando un coagulo in 3 ml di una soluzione di NaCl e un altro coagulo in 3 ml di urea 5M per 24 ore a 37°C. La lisi del coagulo incubato in soluzione di NaCl indica un'eccessiva fibrinolisi. La lisi del coagulo incubato con urea indica un deficit di fattore XIII. Un test normale non esclude un'anomalia della fibrinolisi di lieve entità ma potenzialmente significativo dal punto di vista clinico (p. es., una riduzione del tasso plasmatico di a2-antiplasmina dal 10 al 30% del range normale). Il test di paracoagulazione plasmatica con protammina garantisce lo screening dei monomeri solubili di fibrina nei pazienti con sospetta CID. Un decimo di volume di solfato di protammina all'1% viene mescolato con il plasma, che, dopo una breve incubazione a 37°C, viene esaminato per ricercare la presenza di precipitati filamentosi di fibrina. Un test positivo supporta ulteriormente la diagnosi di CID, ma un risultato negativo non la esclude. Un risultato falsamente positivo può essere causato da difficoltà nel prelievo venoso o di un'inadeguata anticoagulazione del campione di sangue.

53
I prodotti di degradazione della fibrina possono essere misurati con due test. Nel test per il D-dimero, il plasma è aggiunto non diluito e a varie diluizioni a delle particelle di lattice ricoperte di anticorpi monoclonali che reagiscono esclusivamente con derivati della fibrina che contengono D-dimeri, generati quando la fibrina che ha subito legami covalenti crociati è degradata dalla plasmina. Le misture sono osservate per l'agglutinazione delle particelle di lattice. Gli anticorpi non reagiranno con il fibrinogeno, ragione per la quale il test può essere effettuato con del plasma, né con i prodotti di degradazione del fibrinogeno, poiché questi non hanno subito i legami covalenti crociati. Perciò il test è specifico per i prodotti di degradazione della fibrina. Il plasma non diluito delle persone normali darà un test negativo (< 0,25 mg/ml di D-dimero). Il siero normale può contenere piccole quantità (< 10 mg/ml) di residui di prodotti di degradazione del fibrinogeno. La presenza di agglutinazioni a diluizioni del siero di 1:20 indica l'aumentata quantità di prodotti di degradazione della fibrina ( 40 mg/ml).
. Il siero normale può contenere piccole quantità (< 10 mg/ml) di residui di prodotti di degradazione del fibrinogeno. La presenza di agglutinazioni a diluizioni del siero di 1:20 indica l aumentata quantità di prodotti di degradazione della fibrina ( 40 mg/ml)..")
54
Un tempo di lisi dell'euglobulina fa di solito parte dello screening, se si sospetti un'aumentata attività fibrinolitica. Le euglobuline vengono fatte precipitare mediante diluizione e acidificazione del plasma. La frazione euglobulinica, che è relativamente priva di inibitori della fibrinolisi, viene successivamente fatta coagulare con la trombina mentre si misura il tempo impiegato dal coagulo per dissolversi. Un tempo di lisi normale è > 90 min; un tempo più corto indica un incremento dell'attività plasmatica dell'attivatore del plasminogeno (p. es., come in alcuni pazienti con epatopatia avanzata). Anche una ridotta concentrazione di fibrinogeno plasmatico, comportando la dissoluzione di un coagulo più piccolo, può causare un tempo più corto.
. Anche una ridotta concentrazione di fibrinogeno plasmatico, comportando la dissoluzione di un coagulo più piccolo, può causare un tempo più corto.")
55
Difetti della coagulazione

56
EZIOPATOGENESI e EPIDEMOLOGIA
MALATTIA DI vW DEFINIZIONE Malattia emorragica ereditaria, trasmessa in modo autosomico dominante (raramente recessivo), causata da carenza quantitativa o qualitativa del fattore di Von Willebrand EZIOPATOGENESI e EPIDEMOLOGIA Una mutazione genica a livello del cromosoma 12 causa un difetto di sintesi e secrezione del Von Willebrand a livello endoteliale o la produzione di molecole disfunzionali, con conseguente difetto dell’emostasi primaria e riduzione del fattore VIII circolante. Prevalenza 1% della popolazione. I casi che necessitano di cure mediche sono stimati in circa 1/ Colpisce entrambi i sessi.
, causata da carenza quantitativa o qualitativa del fattore di Von Willebrand. EZIOPATOGENESI e EPIDEMOLOGIA. Una mutazione genica a livello del cromosoma 12 causa un difetto di sintesi e secrezione del Von Willebrand a livello endoteliale o la produzione di molecole disfunzionali, con conseguente difetto dell’emostasi primaria e riduzione del fattore VIII circolante. Prevalenza 1% della popolazione. I casi che necessitano di cure mediche sono stimati in circa 1/ Colpisce entrambi i sessi.")
57
FORME CLINICHE Numerose varianti e tipi della malattia, che variano per modalità di ereditarietà, espressione fenotipica, gravità e risposta alla terapia SINTOMATOLOGIA Sintomatologia emorragica molto variabile nel tempo e a seconda del tipo della malattia, a insorgenza precoce nelle forme gravi, tardiva, accidentale o latente nelle lievi. PERCORSO DIAGNOSTICO Dati anamnestici personali e familiari. aPTT allungamento; tempo di emorragia allungato (variabile nelle diverse forme). Aggregazione piastrinica in vitro alla ristocetina diminuita Dosaggio del fattore di Von Willebrand. Tipizzazione: analisi dei multimeri del fattore di Von Willebrand. TERAPIA Desmopressina: efficace in alcuni sottotipi di malattia. Terapia sostitutiva con concentrati plasmatici virus-inattivati di fattore VIII, contenenti anche i multimeri del fattore di Von Willebrand ad alto peso molecolare, nelle forme gravi non responsive alla desmopressina.
. Aggregazione piastrinica in vitro alla ristocetina diminuita Dosaggio del fattore di Von Willebrand. Tipizzazione: analisi dei multimeri del fattore di Von Willebrand. TERAPIA. Desmopressina: efficace in alcuni sottotipi di malattia. Terapia sostitutiva con concentrati plasmatici virus-inattivati di fattore VIII, contenenti anche i multimeri del fattore di Von Willebrand ad alto peso molecolare, nelle forme gravi non responsive alla desmopressina.")
58
Emofilia Malattia ereditaria trasmessa come carattere recessivo legato al cromosoma X, dovuta a deficienza di fattore VIII (emofilia vera o emofilia A) o IX (malattia di Christmas o emofilia B) ETIOPATOGENESI ED EPIDEMIOLOGIA Mutazione sul gene del fattore VIII o IX che induce un difetto di sintesi e conseguente carenza di fattore VIII o IX plasmatico nel maschio, mentre le femmine sono portatrici sane. Prevalenza 1: soggetti di sesso maschile. L’emofilia A è circa 6 volte più frequente dell’emofilia B.
 o IX (malattia di Christmas o emofilia B) ETIOPATOGENESI ED EPIDEMIOLOGIA. Mutazione sul gene del fattore VIII o IX che induce un difetto di sintesi e conseguente carenza di fattore VIII o IX plasmatico nel maschio, mentre le femmine sono portatrici sane. Prevalenza 1: soggetti di sesso maschile. L’emofilia A è circa 6 volte più frequente dell’emofilia B.")
59
FORME CLINICHE grave: attività coagulante del fattore carente<1% moderata: attività coagulante del fattore carente tra 1 e 5% lieve: attività coagulante del fattore carente>5% SINTOMATOLOGIA Emorragie spontanee o post traumatiche prevalentemente articolari, muscolari Complicazioni: artropatia anchilosante da emartri recidivanti. infezioni da virus (epatitici, HIV) trasmessi dagli emoderivati non virus inattivati (precedentemente al 1985). Insorgenza di inibitori anti FVIII/IXi (alloanticorpi) nel 20-35% degli emofilici A gravi.
 trasmessi dagli emoderivati non virus inattivati (precedentemente al 1985). Insorgenza di inibitori anti FVIII/IXi (alloanticorpi) nel 20-35% degli emofilici A gravi.")
60
Percorso diagnostico Diagnosi differenziale
Dati anamnestici personali e familiari. Allungamento dell’aPTT con normale tempo di protrombina, di emorragia e conteggio delle piastrine. L’allungamento dell’aPTT è corretto dal plasma normale; diagnosi confermata con il dosaggio del fattore specifico. Controlli periodici per eventuale sviluppo di inibitore (non correzione dell’aPTT con il plasma normale dopo incubazione a 37° per due ore) Diagnosi di portatrice e prenatale: indagini di biologia molecolare. Diagnosi differenziale Morbo di Von Willebrand (sesso, assenza di emartri, tempo di emorragia, dosaggio del fattore di Von Willebrand) Presenza di inibitori acquisiti (autoanticorpi). Prognosi Severa, migliorata per possibilità di cura della sintomatologia emorragica e delle sue complicanze. Terapia Terapia sostitutiva: concentrati plasmatici virus inattivati o ricombinanti; Emofilia A lieve: desmopressina. Terapia antifibrinolitica nelle emorragie mucose e nella profilassi pre-estrazione dentaria insieme al fattore VIII o IX.
 Diagnosi di portatrice e prenatale: indagini di biologia molecolare. Diagnosi differenziale. Morbo di Von Willebrand (sesso, assenza di emartri, tempo di emorragia, dosaggio del fattore di Von Willebrand) Presenza di inibitori acquisiti (autoanticorpi). Prognosi. Severa, migliorata per possibilità di cura della sintomatologia emorragica e delle sue complicanze. Terapia. Terapia sostitutiva: concentrati plasmatici virus inattivati o ricombinanti; Emofilia A lieve: desmopressina. Terapia antifibrinolitica nelle emorragie mucose e nella profilassi pre-estrazione dentaria insieme al fattore VIII o IX.")
61
ALTERAZIONI ACQUISITE DELLA COAGULAZIONE
DIFETTI DI PRODUZIONE: insufficienze epatiche gravi (7 fattori sono sintetizzati dal fegato tra cui fibrinogeno e protrombina) DIFETTO DI APPROPRIATA MODIFICAZIONE POST-TRADUZIONALE: deficienza di Vitamina K (fattori VII, IX, X) AUMENTATO CONSUMO: Coagulazione Intravascolare Disseminata INATTIVAZIONE: - Ab anti-fattori della coagulazione nei politransfusi e in altre condizioni - terapia con eparina IPERATTIVAZIONE DELLA FIBRINOLISI: - Tumori (rilascio di attivatori del plasminogeno) - Terapie incongrue (streptochinasi)
 DIFETTO DI APPROPRIATA MODIFICAZIONE POST-TRADUZIONALE: deficienza di Vitamina K (fattori VII, IX, X) AUMENTATO CONSUMO: Coagulazione Intravascolare Disseminata. INATTIVAZIONE: - Ab anti-fattori della coagulazione nei politransfusi e in altre condizioni. - terapia con eparina. IPERATTIVAZIONE DELLA FIBRINOLISI: - Tumori (rilascio di attivatori del plasminogeno) - Terapie incongrue (streptochinasi)")
62
DEFICIENZE DI VITAMINA K
SINDROMI DA MALASSORBIMENTO INTESTINALE CON STEATORREA DEFICIT DI FUNZIONE EPATICA CON RIDOTTA SECREZIONE BILIARE INSUFFICIENTE INTRODUZIONE CON LA DIETA (neonati e lattanti) TRATTAMENTO CON FARMACI ANTAGONISTI
 TRATTAMENTO CON FARMACI ANTAGONISTI.")
63
Coagulazione Intravasculare Disseminata (CID)
Dipende dall’attivazione della reazione a cascata della coagulazione con formazione intravasale, cioè dentro i vasi e delle più fine ramificazioni capillari, di microtrombi di fibrina. A tale processo consegue una reazione omeostatica di equilibrio di iperfibrinolisi secondaria.

64
Eziologia a) Immissione in circolo di attivatori della protrombina: -embolia di liquido amniotico aspirato dopo il parto, distacco precoce di placenta, atonia con emorragia post-partuum, aborto settico, ritenzione di feto morto, aborto da cloruro di sodio; -interventi su organi ricchi di trombochinasi, per es. sul polmone, pancreas, prostata, placenta (le 4 P!); -emolisi grave, per es. per incompatibilità in errori trasfusionali; -veleno di serpente; -stati neoplastici terminali con liberazione in circolo di sostanze tromboplastinosimili, nelle leucemie.
; -emolisi grave, per es. per incompatibilità in errori trasfusionali; -veleno di serpente; -stati neoplastici terminali con liberazione in circolo di sostanze tromboplastinosimili, nelle leucemie.")
65
b) Attivazione della coagulazione tramitemediatori:
- endotossine di batteri gram-negativi in gravide; - sindrome di Waterhouse-Friderichsen=coagulopatia da consumo con emorragia cutanea, shoch, rigidità nucale ed emorragie cutanee e surrenali nella sepsi da meningococco à TERAPIA URGENTE CON PENICILLINA G E.V.!! - Coagulopatia da consumo nella setticemia (cioè quando i batteri sono nel sangue) da batteri gram negativi; - Porpora fulminante: affezione acuta, con microtrombi vasale post-infettiva con emorragie cutanee estese e simmetriche della cute e necrosi centrale e DIC.
 da batteri gram negativi; - Porpora fulminante: affezione acuta, con microtrombi vasale post-infettiva con emorragie cutanee estese e simmetriche della cute e necrosi centrale e DIC.")
66
Nella Coagulopatia Intravasale Disseminata o CID o DIC avremo 3 fasi:
fase pre DIC: presenza di malattie e stati a rischio; fase della DIC: alterazioni di laboratorio e diatesi emorragica fase post-DIC: ipercoagulabilità reattiva , ma via via non si rilevano i prodotti di degradazione del fibrinogeno che in un primo momento sono presenti ad alto dosaggio

67
Clinica Il malato presenta shock, con polso piccolo e frequente, agitazione, pallore, sudorazione fredda e labbra cianotiche; possibilmente ha delle petecchie e/o porpora emorragica, cioè delle macchie che sembrano “piccoli nevi puntiformi” diffusi in tutto il corpo, mentre si manifestano emorragie (per es. in un caso clinico una paziente aveva avuto una “ripresa del ciclo”, secondo quanto appreso all’anamnesi, emissione di sangue nelle urine, lacrime miste a sangue, insufficienza cardio-respiratoria acuta, fino al coma irreversibile ed all’exitus, con consumo delle piastrine, aumento dei PDF e del D-dimero Diagnosi E’ la fase più delicata della CID e presuppone che un medico abbia il sospetto clinico di ciò che cerca , che presuppone l’attenta ed umile valutazione del malato.

68
LABORATORIO: all’emocromo avremo calo continuo delle piastrine, anche nel giro di minuti!! fibrinogeno ed ANTI TROMBINA III che si consumano e si riducono velocemente; dimostrazione di monomeri di fibrina; Prodotti di degradazione del Fibrinogeno D-Dimero (!!!!) Tempo di Quick che si riduce; PTT che aumenta Riduzione dei fattori V = Trombina Riduzione dei fattoti VIII
 Tempo di Quick che si riduce; PTT che aumenta. Riduzione dei fattori V = Trombina. Riduzione dei fattoti VIII.")
69
ANTICOAGULANTI La coagulazione viene ritardata da: bassa temperatura,
aumento PCO2, contatto del sangue con superfici non bagnabili. Vi sono sostanze che sono in grado di opporsi alla coagulazione del sangue. Gli anticoagulanti vengono usati sistematicamente in un certo numero di malattie o di stati gravi di cui migliorano la prognosi. Malattie in cui vengono utilizzati: Trombosi in cui vi è coagulazione del sangue seguita da un’ostruzione e temibili complicazioni; Arteriosclerosi; Flebiti; Malattie del cuore(per esempio in seguito a stenosi mitralica, fibrillazione atriale).
.")
70
EPARINA Una potente azione anticoagulante è esplicata dall’ EPARINA, sostanza estratta dal fegato, ma ottenuta anche da altri organi come il polmone e la mucosa intestinale. Granuli di eparina si trovano nelle mastcellule. Questi granuli vengono colorati metacromaticamente in porpora di bleu di toluidina. Chimicamente l’eparina è un polisaccaride costituito da glusosamina e acido glicuronico e pertanto contiene nella sua molecola numerosi gruppi SO4. E’ fornita di un’energica carica negativa che sarebbe responsabile della sua attività anticoagulante. Essa interviene in tutti e tre gli stadi della coagulazione formando dei complessi inattivi di AT-III con la trombina e le altre serinproteasi.

71
Un’altra sostanza che inibisce la coagulazione del sangue è il DICUMAROLO: chimicamente è un derivato del naftochinone. Agisce in vivo, ma non in vitro. Se somministrato all’organismo vivente esplica un’azione antivitamina K e determina diminuzione del livello ematico di protrombina e del fattore VII. Si ritiene che il dicumarolo antagonizzi la vitamina K prendendo nel fegato il posto e blocchi la sintesi dei fattori II, VII, IX e X. Un anticoagulante che agisce come antivitamina K è pure il FENINDIONE.

Presentazioni simili













.>")


 Costituito da una parte liquida: plasma e.>")


>")






 Le cellule distanti tra loro comunicano attraverso molecole (MEDIATORI o NEUROTRASMETTITORI)>")
