Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Fibrosi cistica Prof. Carlo Vancheri
DIPARTIMENTO DI MEDICINA INTERNA E SPECIALISTICA SEZIONE DI MALATTIE DELL’APPARATO RESPIRATORIO UNIVERSITA’ DI CATANIA Fibrosi cistica Prof. Carlo Vancheri

2
Fibrosi cistica Definizione:
La Fibrosi Cistica, detta anche mucoviscidosi, è una malattia genetica letale ad ereditarietà autosomica recessiva che colpisce le ghiandole a secrezione mucosa ed è caratterizzata da abnorme accumulo di muco vischioso (da cui mucoviscidosi), con ostruzione e dilatazione dei dotti ghiandolari. Si presenta come malattia sistemica con alterazioni a carico delle vie aeree, insufficienza pancreatica esocrina, anomala funzione delle ghiandole sudoripare, disfunzioni epatiche e della sfera genitale.
, con ostruzione e dilatazione dei dotti ghiandolari. Si presenta come malattia sistemica con alterazioni a carico delle vie aeree, insufficienza pancreatica esocrina, anomala funzione delle ghiandole sudoripare, disfunzioni epatiche e della sfera genitale.")
3
Fibrosi cistica ( CF) Epidemiologia:
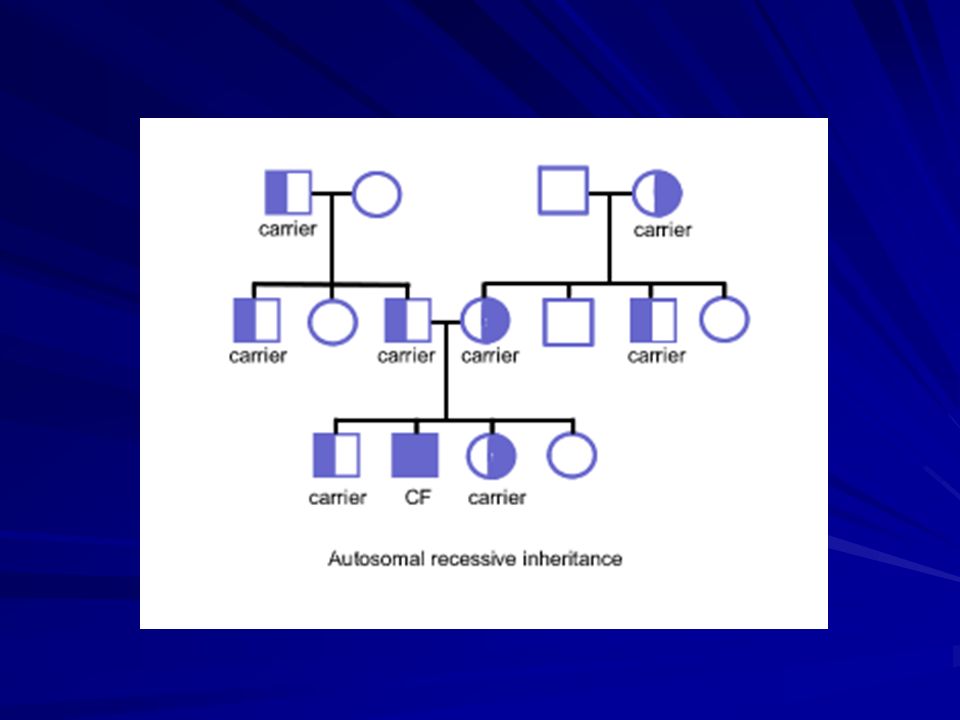
La CF è la più frequente malattia genetica letale nella razza bianca. La malattia è meno frequente nella razza nera e nelle popolazioni asiatiche. Incidenza: 1/2500 nati vivi; Trasmissione: autosomica recessiva; Rapporto M/F= 1/1; Si calcola che circa il 5% della popolazione sia costituita da eterozigoti, i cosiddetti portatori sani.

5
Fibrosi cistica ( CF) Eziologia:
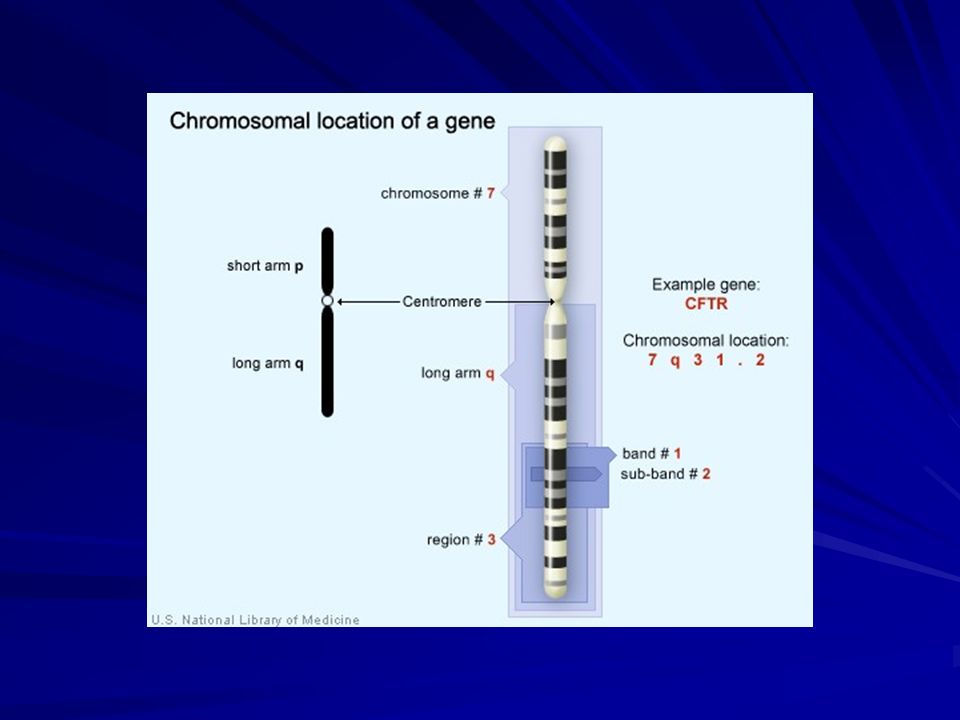
La CF è una malattia autosomica recessiva che consegue a mutazioni di un gene (FC), localizzato entro circa 300 Kb nella regione q21-31 del cromosoma 7. Composto da 27 esoni separati da 26 introni, esso codifica per una proteina, la Cystic Fibrosis Transmembrane Regulator ( CFTR) , costituita dalla sequenza nucleotidica di 1480 amminoacidi, e la cui funzione è quella di regolare il trasporto del cloro.
, localizzato entro circa 300 Kb nella regione q21-31 del cromosoma 7. Composto da 27 esoni separati da 26 introni, esso codifica per una proteina, la Cystic Fibrosis Transmembrane Regulator ( CFTR) , costituita dalla sequenza nucleotidica di 1480 amminoacidi, e la cui funzione è quella di regolare il trasporto del cloro.")
9
Fibrosi cistica ( CF) Eziologia:
Gene CF ( coppie di basi) Proteina CFTR ( 1480 aa) ; Le mutazioni genetiche già identificate sono circa 400; La loro frequenza varia in base al substrato etnico e geografico della popolazione ( ad esempio W1282X = 50% di CF nella razza giudaica). Nel 75% dei casi di CF la mutazione genica determina una delezione ( Δ ) di un residuo di Fenilalanina (F) in posizione 508 della catena aminoacidica.
 Proteina CFTR ( 1480 aa) ; Le mutazioni genetiche già identificate sono circa 400; La loro frequenza varia in base al substrato etnico e geografico della popolazione ( ad esempio W1282X = 50% di CF nella razza giudaica). Nel 75% dei casi di CF la mutazione genica determina una delezione ( Δ ) di un residuo di Fenilalanina (F) in posizione 508 della catena aminoacidica.")
10
Fibrosi cistica ( CF) Eziologia:
Mutazione: CF gene ( coppie di basi) -3 coppie di basi = CFTR (1480 aa)– 1 amminoacido = 1479 aa. ΔF 508 : ( 70-75% ) Δ = Deletion F = Fenilalanina
 -3 coppie di basi = CFTR (1480 aa)– 1 amminoacido = 1479 aa. ΔF 508 : ( 70-75% ) Δ = Deletion. F = Fenilalanina.")
11
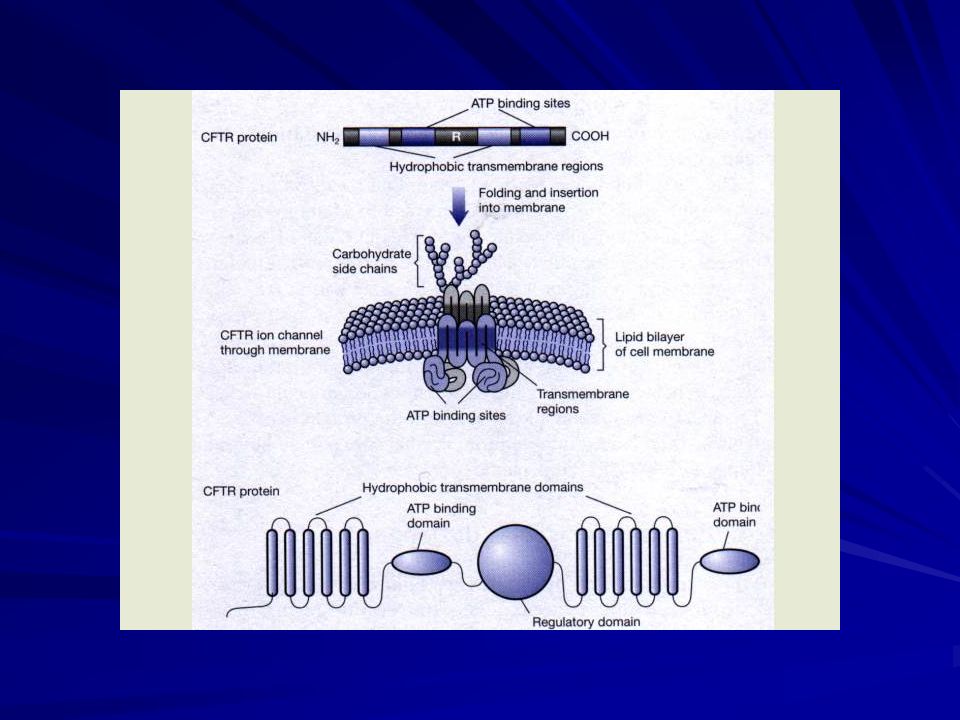
Struttura della CFTR: Consiste di una porzione che attraversa la membrana cellulare con 12 α-eliche e di 3 regioni intracitoplasmatiche, di cui: -2 regioni globulari che legano 1 molecola di ATP ciascuna ( Nucleotide Binding Folds o NBF); -1 regione regolatrice ( R) che contiene numerosi siti di fosforilazione per PKA e PKC.
; -1 regione regolatrice ( R) che contiene numerosi siti di fosforilazione per PKA e PKC.")
12
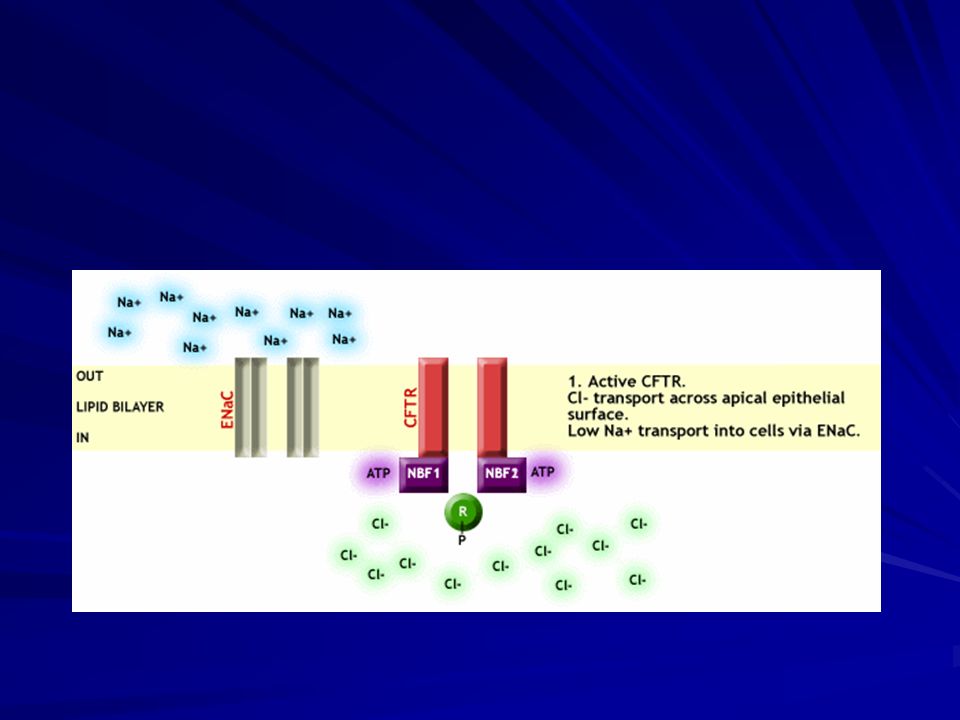
Fibrosi cistica Azione svolte dal CFTR:
Regolazione del passaggio degli ioni Cl Regolazione di altri canali al Cloro Regolazione dei canali del Sodio

21
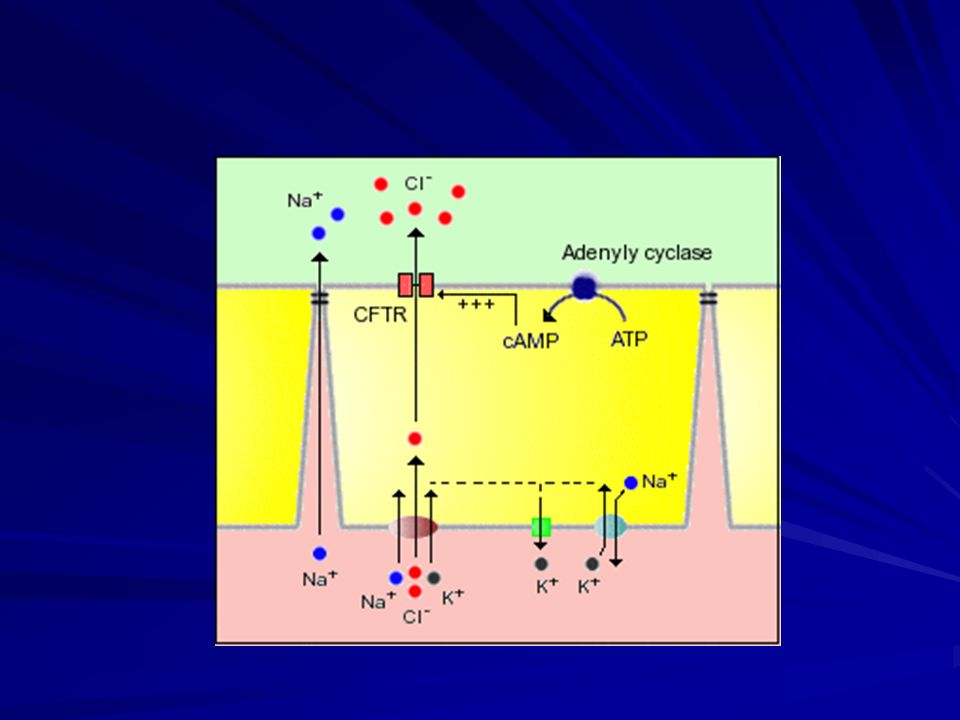
Fibrosi cistica Struttura della CFTR:
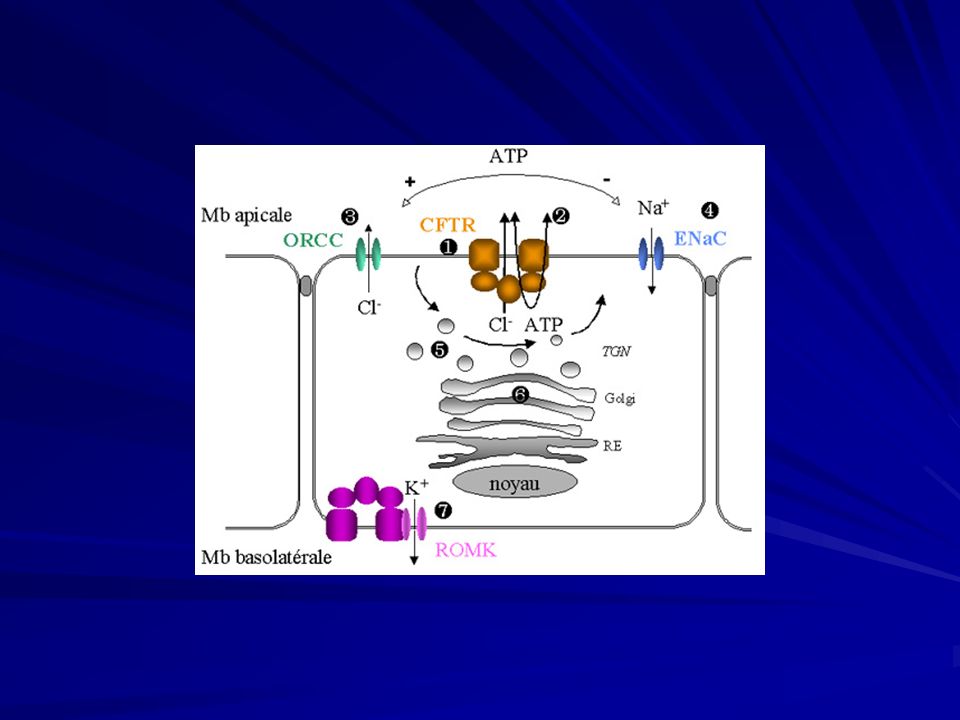
La proteina CFTR funziona come un canale di secrezione di ioni cloro, regolato dal cAMP, sulla superficie apicale delle cellule epiteliali. Cl NBF 1 Cl Cl ATP Cl R Cl cAMP pKA Cl PO4 Cl Cl NBF 2 Cl ATP REGIONE TRANSMEMBRANA

22
Fibrosi cistica Metabolismo intracellulare della CFTR:
La CFTR viene codificata nel nucleo, sintetizzata nel reticolo endoplasmatico rugoso ( RER) , glicosilata nel complesso di Golgi e trasportata infine sulla membrana plasmatica. LUME CELL NORMALE CL cAMP GOLGI RER SANGUE NUCLEO
 , glicosilata nel complesso di Golgi e trasportata infine sulla membrana plasmatica. LUME. CELL NORMALE. CL. cAMP. GOLGI. RER. SANGUE. NUCLEO.")
23
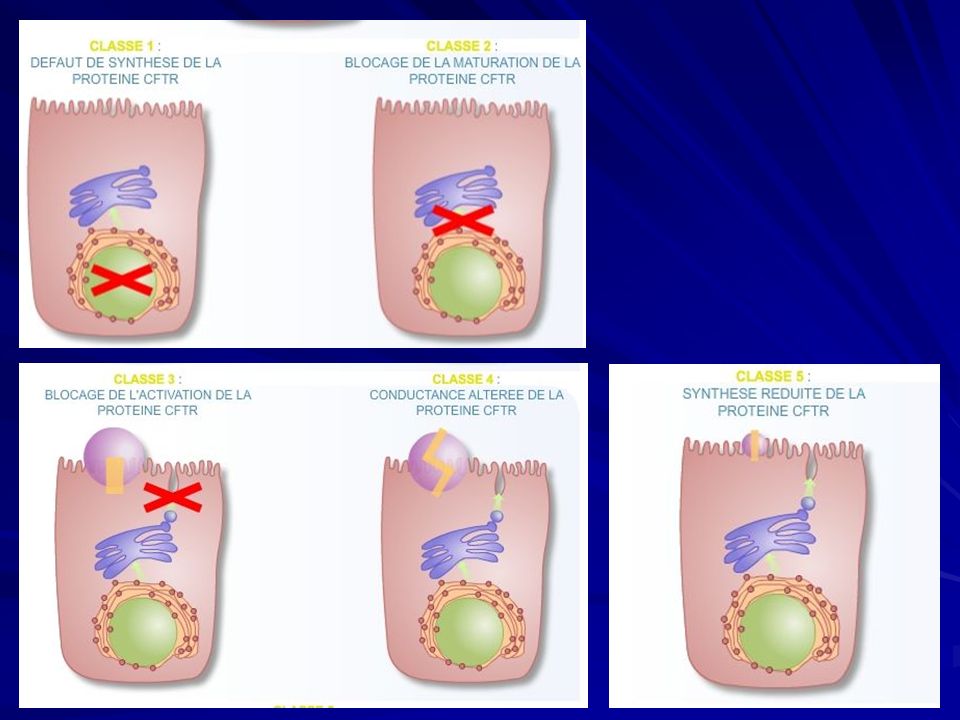
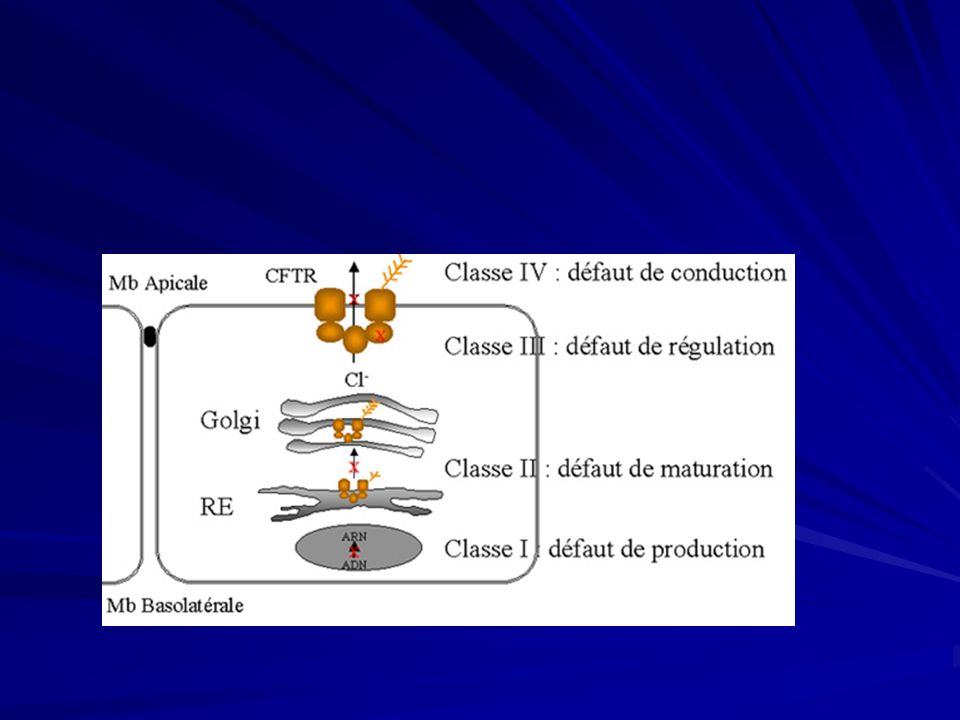
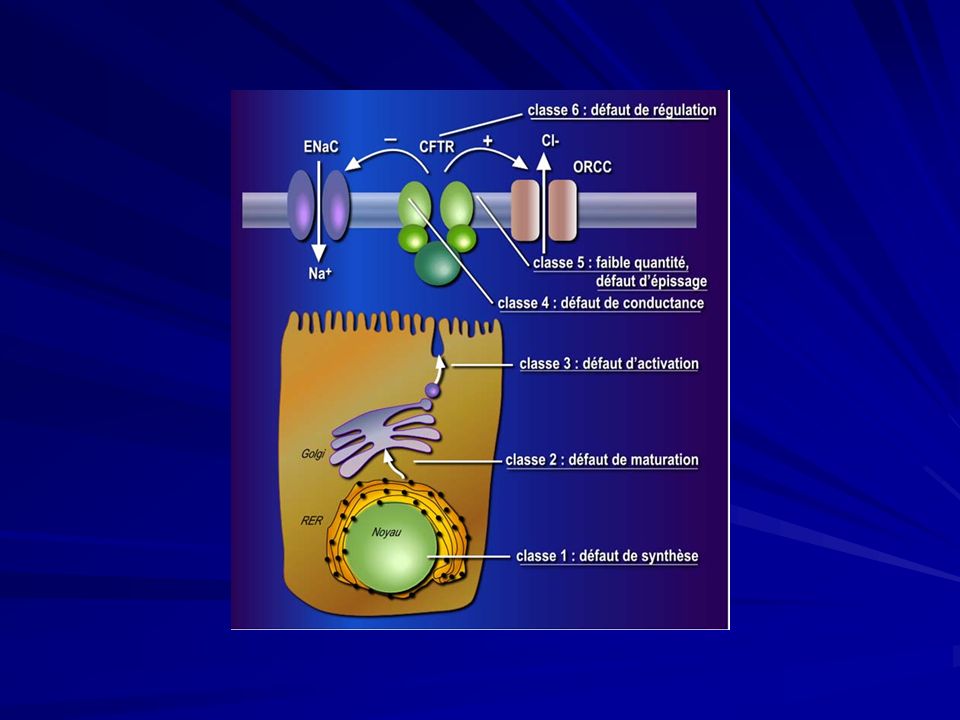
Fibrosi cistica Classi di mutazione della proteine CFTR:
Difettosa glicosilazione della proteina CFTR, aumentata affinità per il RER con la relativa degradazione intracellulare ( ΔF 508); 2) La proteina CFTR anomala viene portata alla membrana plasmatica, ma non risponde ai segnali di attivazione per mancata fosforilazione ( G551); 3) Arresto prematuro della traslazione dell’mRNA del CFTR (G542); 4) Normale presenza sulla membrana cellulare ma funzionamento anomalo per alterazioni nella dimensione del canale o del tempo di apertura ( R117).
; 2) La proteina CFTR anomala viene portata alla membrana plasmatica, ma non risponde ai segnali di attivazione per mancata fosforilazione ( G551); 3) Arresto prematuro della traslazione dell’mRNA del CFTR. (G542); 4) Normale presenza sulla membrana cellulare ma funzionamento anomalo per alterazioni nella dimensione del canale o del tempo di apertura ( R117).")
26
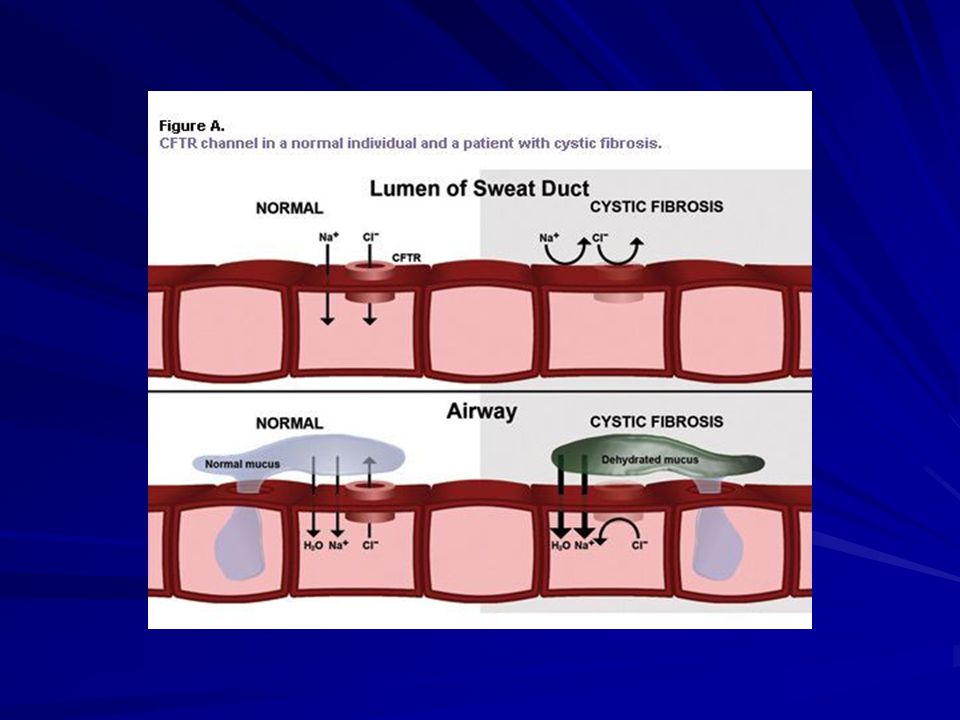
Fibrosi cistica L’alterazione della CFTR determina:
Diminuita secrezione del cloro in modo diretto ed indiretto; Aumentato assorbimento del sodio in modo attivo e passivo; Alterata composizione delle mucine; Alterazione del ruolo difensivo del CFTR; Alterazione delle β-defensine e catelecidine.

27
Fibrosi cistica CFTR e vie aeree:
-Ipotesi del basso volume ( low volume hypothesis) -Ipotesi dell’eccesso di sale (high salt hypothesis)
 -Ipotesi dell’eccesso di sale (high salt hypothesis)")
28
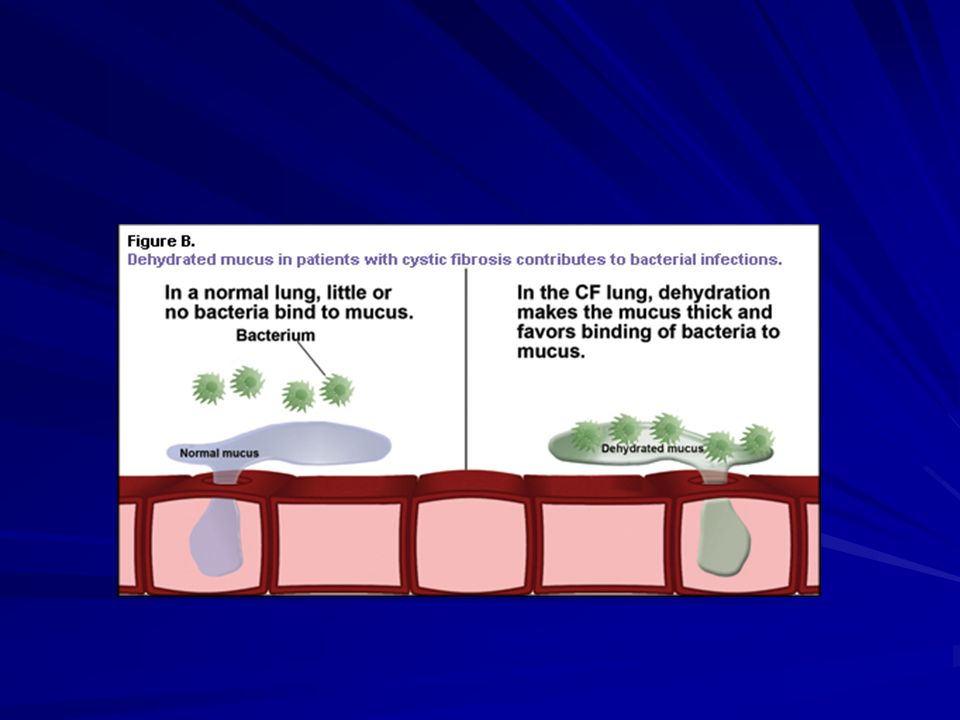
Fibrosi cistica Patogenesi del danno polmonare nella CF
Proteina CFTR alterata Assorbimento Cl- Secrezione Cl- Assorbimento Na+ Assorbimento H2O Contenuto in H2O, Cl- e Na+ nelle secrezioni bronchiali Disidratazione delle secrezioni Viscosità del muco Clearance mucociliare Infiammazione Infezione

32
Fibrosi cistica I neutrofili, attraverso la produzione di Elastasi, svolgono un ruolo centrale nel determinare la flogosi cronica dell’epitelio bronchiale. CR1, C3bi cleavage LTB4, IL8 IL8 epiteliale NEUTROFILI ELASTASI Deficit di opsonizzazione DNA secrezione IgG cleavage O2-, H2O2 Formazione di plugs Degradazione elastina DANNO EPITELIALE, BRONCHIECTASIE

33
RIACUTIZZAZIONI POLMONARI RISPOSTA INFIAMMATORIA
Fibrosi cistica FIBROSI CISTICA ALTERAZIONI MUCO INFEZIONI CRONICHE RIACUTIZZAZIONI POLMONARI RISPOSTA INFIAMMATORIA OSTRUZIONE VIE AEREE BRONCHIECTASIE ATELECTASIA PNM- EMOTTISI ALTERAZIONE VA/Q IPOSSIA IPERCAPNIA CUORE POLMONARE INSUFF RESPIRAT

34
Fibrosi cistica CUORE POLMONARE CRONICO INSUFFICIENZA RESPIRATORIA
FLOGOSI CRONICA OSTRUZIONE BRONCHIALE GRAVE ENFISEMA COMPENSATORIO ATELETTASIA ALTERAZIONI VA/Q IPOSSIA CRONICA IPERCAPNIA CUORE POLMONARE CRONICO INSUFFICIENZA RESPIRATORIA

35
Fibrosi cistica Infezioni nella fibrosi cistica:
-Batteriche: Pseudomonas aeruginosa Burkholderia cepacia Stafilococcus Aureus Haemofilus influenzae -Virali: Virus influenzali Virus respiratorio sinciziale -Micotiche: Aspergillus fumigatus Candida -Micobatteri Non tubercolari: M. Avium intracellulare M. Chelonei

37
Dal gene CFTR alle secrezioni dense
La Fibrosi Cistica Dal gene CFTR alle secrezioni dense Secrezioni vischiose e difficili da eliminare: polmoni, seni paranasali, pancreas, intestino, ghiandole sudoripare, fegato apparato riproduttivo. Danni funzionali da ristagno: Ostruzione infezione

38
Coinvolgimento di organi extrapolmonari nella CF:
PANCREAS ( Frequenza 85%) Ostruzione dei dotti pancreatici ad opera del secreto abnorme e vischioso cui conseguono: Dilatazione degli acini ghiandolari Degenerazione del parenchima in fibrosi e atrofia. La fibrosi può coinvolgere anche le isole di langherans con ridotta tolleranza glucidica o diabete conclamato. Le alterazioni del succo pancreatico consistono in una riduzione del volume di secreto e un deficit di bicarbonato e dei principali enzimi: Tripsina, chimotripsina, lipasi, amilasi Sindrome maldigestiva.
 Ostruzione dei dotti pancreatici ad opera del secreto abnorme e vischioso cui conseguono: Dilatazione degli acini ghiandolari. Degenerazione del parenchima in fibrosi e atrofia. La fibrosi può coinvolgere anche le isole di langherans con ridotta tolleranza glucidica o diabete conclamato. Le alterazioni del succo pancreatico consistono in una riduzione del volume di secreto e un deficit di bicarbonato e dei principali enzimi: Tripsina, chimotripsina, lipasi, amilasi Sindrome maldigestiva.")
39
Coinvolgimento di organi extrapolmonari nella CF:
Fegato e vie biliari: (Frequenza 50%) La bile, densa e vischiosa, ostruisce le vie biliari intraepatiche producendo ittero di tipo misto e reazioni infiammatorie del parenchima che sfociano in CIRROSI BILIARE. La colecisti può contenere dei calcoli biliari.
 La bile, densa e vischiosa, ostruisce le vie biliari intraepatiche producendo ittero di tipo misto e reazioni infiammatorie del parenchima che sfociano in CIRROSI BILIARE. La colecisti può contenere dei calcoli biliari.")
40
Coinvolgimento di organi extrapolmonari nella CF:
GHIANDOLE SUDORIPARE (Frequenza 98%) Esse secernono volumi normali di sudore nell’acino sudoriparo, ma non sono in grado di riassorbire il NaCl dal sudore mentre fluisce lungo il dotto escretore poiché l’epitelio duttale, nella CF, è impermeabile al cloro. Sudore eccessivamente salato con Cl-, Na+, K+ Cloro nel sudore> 60 mEq/L
 Esse secernono volumi normali di sudore nell’acino sudoriparo, ma non sono in grado di riassorbire il NaCl dal sudore mentre fluisce lungo il dotto escretore poiché l’epitelio duttale, nella CF, è impermeabile al cloro. Sudore eccessivamente salato con. Cl-, Na+, K+ Cloro nel sudore> 60 mEq/L.")
41
Coinvolgimento di organi extrapolmonari nella CF:
NASO E SENI PARANASALI (85-100%) -Poliposi nasale e mucocele ( raccolta di muco denso nei seni paranasali). -Sinusite cronica con frequenti riacutizzazioni sostenute da Pseudomonas, H. Influenzae, Streptococchi e anaerobi. -Anosmia.
 -Poliposi nasale e mucocele ( raccolta di muco denso nei seni paranasali). -Sinusite cronica con frequenti riacutizzazioni sostenute da Pseudomonas, H. Influenzae, Streptococchi e anaerobi. -Anosmia.")
42
Coinvolgimento di organi extrapolmonari nella CF:
INTESTINO ( frequenza 50%) La riduzione del contenuto in acqua delle feci determina ileo da meconio nel neonato ed equivalenti patologie ostruttive nell’adulto.
 La riduzione del contenuto in acqua delle feci determina ileo da meconio nel neonato ed equivalenti patologie ostruttive nell’adulto.")
43
Coinvolgimento di organi extrapolmonari nella CF:
ORGANI SESSUALI (Frequenza 90%) I maschi adulti presentano sterilità da azospermia e da alterazioni del succo prostatico. Le donne presentano un muco cervicale denso che ne riduce la fertilità.
 I maschi adulti presentano sterilità da azospermia e da alterazioni del succo prostatico. Le donne presentano un muco cervicale denso che ne riduce la fertilità.")
44
CLINICA: ILEO DA MECONIO:
Alla nascita, dovuto in parte all’assenza di amilasi pancreatica ed in parte all’alterata secrezione gastrointestinale di muco. SINTOMATOLOGIA RESPIRATORIA: Insorge nelle prime settimane o nei primi mesi di vita con: -tosse, inizialmente secca , poi produttiva -Dispnea fischiante -Episodi bronchitici frequenti -Possibile esordio con pneumotorace od emoftoe.

45
CLINICA: SINTOMATOLOGIA MALDIGESTIVA:
-Scarsissimo accrescimento alla nascita; -Diarrea con feci voluminose, untuose, contenenti grassi neutri non digeriti, proteine ed amidi-Addome globoso; -Deficit di vitamine liposolubili: Vit. D Rachitismo; Vit K emorragie -Deficit di minerali: Ca osteoporosi. SINTOMATOLOGIA EPATICA: Ittero prolungato di tipo misto

46
DIAGNOSI Criteri maggiori: -Cl- sudore > 60mEq/l e
-Malattia polmonare cronica ostruttiva con infezione batterica e/o -Insuffivienza pancreatica e/o -Familiarità positiva per FC Criteri minori: -Azoospermia inspiegata; -Presenza di Pseudomonas nell’esame colturale dell’espettorato.

47
Test diagnostici -Test del sudore; -Esame delle feci;
-Test della secretina-pancreozimina; -Test di funzionalità respiratoria.

48
Indagini strumentali RX del torace; Tomografia computerizzata;
ANALISI DEL DNA

49
Valutazione del danno polmonare nella fibrosi cistica
RX e TC torace Atelettasie Bronchiectasie Lesioni cistiche Test di funzionalità Polmonare FEV1, VR EGA PaO2, PaCO2

50
Cascata fisiopatologica e terapia
Gene anomalo Terapia genica CFTR anomalo Terapia sostitutiva Cl- Na Amiloride, ATP Secrezioni vischiose DNAsi, mucolitici Ostruzione bronchiale Broncodilatatori Fisioterapia resp. Infezioni Antibioticoterapia Infiammazione Steroidi, FANS, Antiproteasi, antiossidanti Bronchiectasie DNAsi, fisioterapia resp. Insufficienza resp Trapianto polmonare

51
Terapia genica e sostitutiva
Proteina CFTR: Liposomi Virosomi cDNA CFTR: Adenovirus Parvovirus Liposomi

52
Criteri di selezione per il trapianto polmonare nella Fibrosi cistica:
INDICAZIONI: IRC nonostante trattamento medico Grave decadimento della qualità di vita Forte motivazione da parte del paziente CONTROINDICAZIONI: Alte dosi di steroidi Instabilità psico-sociale Infezioni da Mycobacterium o Aspergillus Grave interessamento di altri organi Grave malnutrizione FATTORI DI RISCHIO: Precedente chirurgia toracica Ventilazione meccanica Grave insuff. Epatica Diabete Infezione da BurKholderia cepacea

54
La Fibrosi Cistica Test diagnostici
Misura del cloruro di sodio nel sudore Misura dei grassi fecali e degli enzimi pancreatici Esami strumentali: TAC polmonare (bronchiectasie ?) Ecografia addominale (epatobiliare) TAC seni paranasali (ispessimento della mucosa) Test genetici: - Mutazione più frequente DF508
 Ecografia addominale (epatobiliare) TAC seni paranasali (ispessimento della mucosa) Test genetici: - Mutazione più frequente DF508.")
55
La Fibrosi Cistica CFTR Canale del cloro DF508
Presente nel 90% dei casi Condiziona una più breve emivita (rapida degradazione) della proteina Per alterato folding …
 della proteina. Per alterato folding …")
57
La Fibrosi Cistica … - Ileo da meconio
Prolungato ittero neonatale (bile ispessita) Manifestazioni polmonari Successivamente evidente: ritardo di crescita Infezioni respiratorie ricorrenti (anche polmoniti) Tosse cronica Colonizzazione cronica con specifici batteri patogeni: Pseudomonas aeruginosa Burkholderia (Pseudomonas) cepacia Maldigestione con: dolori addominali meteorismo feci abbondanti e ricche di grassi - Stasi biliare Neonato Bambino
 Manifestazioni polmonari. Successivamente evidente: ritardo di crescita. Infezioni respiratorie ricorrenti (anche polmoniti) Tosse cronica. Colonizzazione cronica con specifici batteri patogeni: Pseudomonas aeruginosa. Burkholderia (Pseudomonas) cepacia. Maldigestione con: dolori addominali meteorismo. feci abbondanti e ricche di grassi. - Stasi biliare. Neonato. Bambino.")
58
La Fibrosi Cistica Terapia Dieta
Ad alto contenuto calorico (diminuito assorbimento aumentato consumo) Con integratori Ricco apporto di liquidi e sali minerali Enzimi pancreatici Terapia antimicrobica - antibiotici - mucolitici Fisioterapia Trapianto di polmone Terapia genica
 Con integratori. Ricco apporto di liquidi e sali minerali. Enzimi pancreatici. Terapia antimicrobica. - antibiotici. - mucolitici. Fisioterapia. Trapianto di polmone. Terapia genica.")
Presentazioni simili