Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Patologia Extracellulare

2
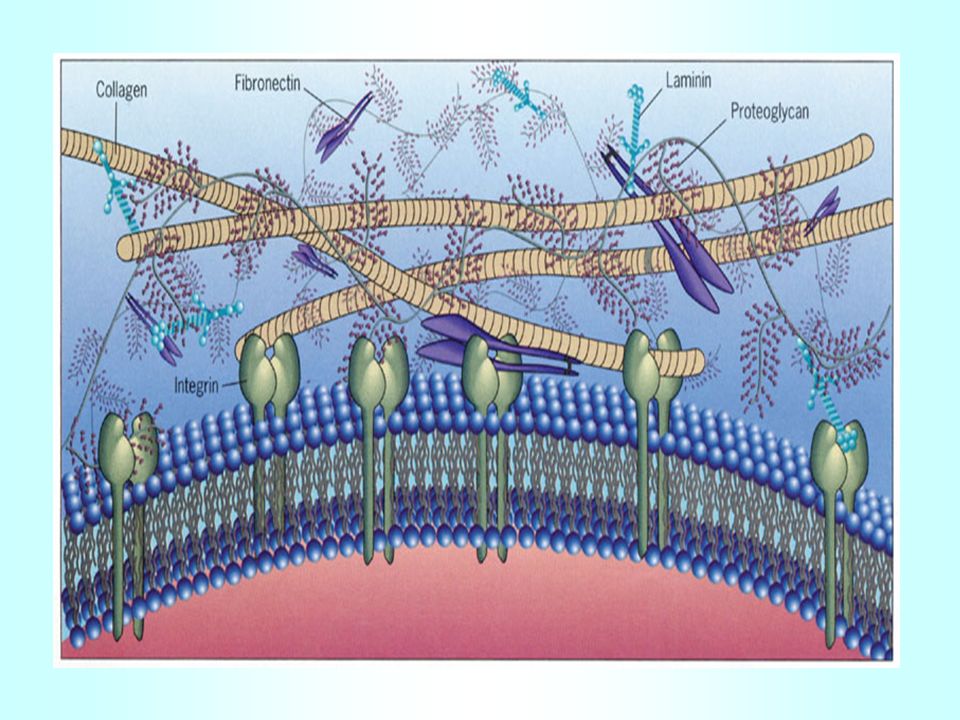
Le cellule aderiscono alle matrici extracellulari mediante recettori per il collageno, lamininina, fibronectina etc, per cui i segnali vengono trasmessi dalla matrice alla cellula Fibronectina Laminina

3
I recettori della famiglia delle integrine collegano le molecole
Stretto rapporto tra cellule ed ambiente esterno. La matrice extracellulare può Influenzare adesione, forma, Movimento ed espressione genica cellulare I recettori della famiglia delle integrine collegano le molecole extracellulari con il reticolo citoscheletrico

4
La matrice extracellulare
Membrana basale Tessuto connettivo Matrice provvisoria Servono all’organizzazione, alle proprietà fisiche e alla funzione dei tessuti

5
Componenti dei tessuti connettivali lassi
Fibre collagene Fibre elastiche Proteoglicani Membrane basali

6
La patologia della matrice extracellulare:
Comprende modificazioni quantitative e qualitative dei suoi componenti ma anche la comparsa di materiali anomali come Ac. Ialuronico, C3, lipidi Le masse ialine possono essere costituite da fibrina, collageno, mucopolisaccaridi o amiloide

7
Patologia del collageno
E’ la proteina più abbondante Rappresenta 1/3 delle proteine dell’organismo Funzione: Garantire l’integrità dell’organismo e di riparare le lesioni Le anomalie conclamate generalizzate sono rare Possono essere congenite, nutrizionali, tossiche Le patologie localizzate sono frequenti e riguardano la riparazione delle ferite

8
Struttura del collageno
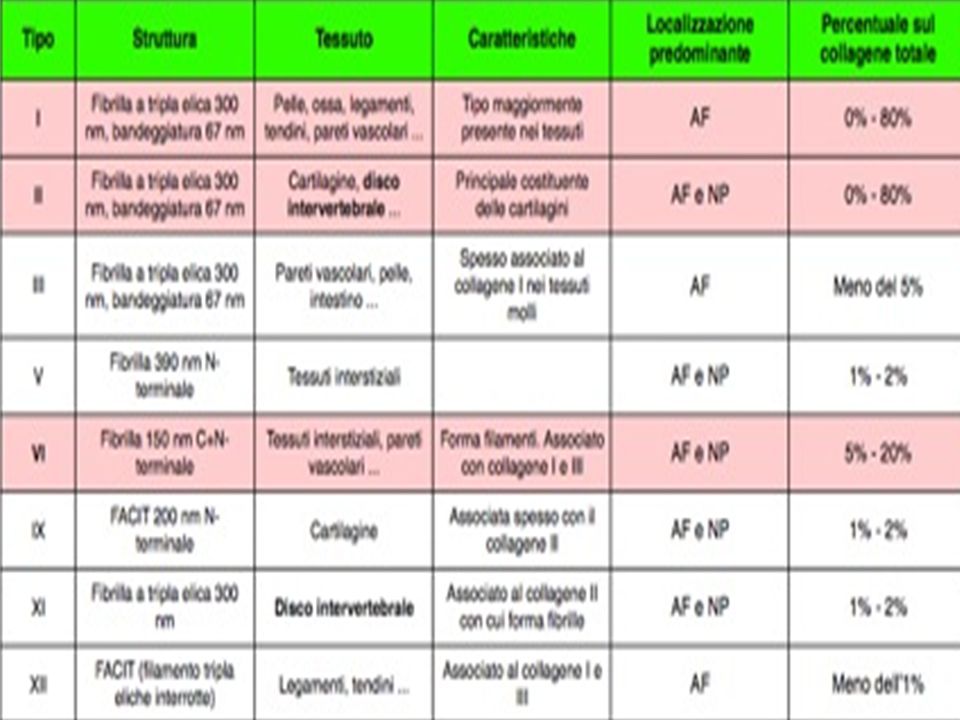
20 tipi di collageno Ciascuna molecola è costituita da tre catene alfa avvolte tra loro come in una fune Sono stati identificate 38 diverse tipi di catene polipeptidiche

9
Collageni Le varietà fibrillari di collageno conferiscono resistenza ai tessuti I tipo: è il principale collageno fibrillare, da forza tensile ai tessuti (cornea e tendini) II tipo: si trova nella cartilagine ialina delle articolazioni III tipo: è formato da fibrille più sottili e sostiene gli organi dilatabili (vasi sanguigni) Varietà non fibrillari IV tipo:. Le sue molecole formano tetrameri a X. Costituisce lo scheletro delle membrane basali I tipi V, VI, IX e XII rivestono le altre fibre collagene
 II tipo: si trova nella cartilagine ialina delle articolazioni. III tipo: è formato da fibrille più sottili e sostiene gli organi dilatabili (vasi sanguigni) Varietà non fibrillari. IV tipo:. Le sue molecole formano tetrameri a X. Costituisce lo scheletro delle membrane basali. I tipi V, VI, IX e XII rivestono le altre fibre collagene.")
10
Interactions of fibrous and nonfibrous collagens
Interactions of fibrous and nonfibrous collagens. (a) Association of types II and IX collagen in a cartilage matrix. Type II forms fibrils similar in structure to type I, with a similar 67-nm periodicity, though smaller in diameter. Type IX contains two long triple helices connected at a flexible kink. At this point a chondroitin sulfate chain is linked to the α2(IX) chain. Type IX collagens are bound at regular intervals along type II fibrils, with an N-terminal nonhelical domain of type IX projecting outward. It is thought that these domains bind the collagen fibrils to the proteoglycan-rich matrix. (b) Organization of the major fibrous components in the extracellular matrix of tendons. Type I fibrils, with their characteristic 67-nm period, are all oriented longitudinally, that is, in the direction of the stress applied to the tendon. The fibrils are coated with an array of proteoglycans, as shown in blue on the right-hand fibril. Type VI fibrils bind to and link together the type I fibrils. Type VI collagen consists of thin triple helices, about 60 nm long, with globular domains at either end. The globular domains of several type VI molecules bind together, giving a “beads-on-a-string” appearance to the type VI fibril Associazione di collageno tipo II e tipo IX in una matrice cartilaginea Organizzazione della maggior parte delle fibre componenti della ME dei tendini
 Association of types II and IX collagen in a cartilage matrix. Type II forms fibrils similar in structure to type I, with a similar 67-nm periodicity, though smaller in diameter. Type IX contains two long triple helices connected at a flexible kink. At this point a chondroitin sulfate chain is linked to the α2(IX) chain. Type IX collagens are bound at regular intervals along type II fibrils, with an N-terminal nonhelical domain of type IX projecting outward. It is thought that these domains bind the collagen fibrils to the proteoglycan-rich matrix. (b) Organization of the major fibrous components in the extracellular matrix of tendons. Type I fibrils, with their characteristic 67-nm period, are all oriented longitudinally, that is, in the direction of the stress applied to the tendon. The fibrils are coated with an array of proteoglycans, as shown in blue on the right-hand fibril. Type VI fibrils bind to and link together the type I fibrils. Type VI collagen consists of thin triple helices, about 60 nm long, with globular domains at either end. The globular domains of several type VI molecules bind together, giving a beads-on-a-string appearance to the type VI fibril. Associazione di collageno tipo II e tipo IX in una matrice cartilaginea. Organizzazione della maggior parte delle fibre componenti della ME dei tendini.")
12
Cellule che secernono collageno:
Fibroblasti Cellule muscolari lisce Cellule epiteliali

13
Deaminazione ossidativa di Lys e Hyl
La sintesi avviene tramite la secrezione di piccoli precursori in grado di auto-assemblarsi e coinvolge 13 enzimi ed un numero doppio di cofattori Idrossilazione Glicosilazione Transferasi Procollageno Procollageno L’mRNA che codifica il collageno emerge dal nucleo e viene tradotto dai ribosomi in 2 tipi di catene pro-alpha recanti alle estremità polipeptidi terminali cruciali per l’assemblaggio, quindi la molecola immatura avanzando attraverso il RE va incontro a processi di idrossilazione e glicosilazione. A questo punto le catene si avvolgono in gruppi di tre a formare un’elica il procollageno. All’esterno della cellula la molecola si auto-assembla non appena i polipeptidi terminali sono rimossi dalle peptidasi extracellulari e si forma il tropocollageno che si assemblano in microfibrille con legami covalenti (cross-linking) Aggregazione delle triple eliche Cross-linking Lisil-ossidasi Deaminazione ossidativa di Lys e Hyl L’intero processo è soggetto ad errori Alcuni avvengono all’interno della cellula, altri riguardano la fibra che si trova all’esterno
 Aggregazione delle triple eliche. Cross-linking. Lisil-ossidasi. Deaminazione ossidativa di Lys e Hyl. L’intero processo è soggetto ad errori. Alcuni avvengono all’interno della. cellula, altri riguardano la fibra che si. trova all’esterno.")
14
Il successo della sintesi del collagene dipende da una serie di modificazioni Post-traduzionali:
Allineanento delle 3 catene Formazione della tripla elica Clivaggio di peptidi terminali non di natura collagene Legame crociato covalente mediato dalla lisil ossidasi Cu dipendente

15
Difetti Genetici Le malattie congenite possono originarsi da difetti:
in geni che codificano il collageno in geni che codificano gli enzimi attivi negli eventi post-traduzionali Sintomi a carico degli organi le cui funzioni meccaniche dipendono dal collageno: Ossa Articolazioni Cute Grosse arterie Valvola mitralica Cristallino

16
Osteogenesis imperfecta Sindrome di Ehler-Danlos
Malattie congenite Osteogenesis imperfecta Sindrome di Ehler-Danlos I difetti delle catene polipeptidiche del collageno la rendono una proteina “strutturalmente incompetente” che viene demolita rapidamente all’interno ed all’esterno della cellula

17
Osteogenesis-imperfecta (Osteopsatirosi o malattia di Lobstein tipo I
Sindrome di Vrolik tipo II°, Sindrome di van der Hoeve, Sindrome di Eddowes) A trasmissione autosomica dominante Mutazioni in uno dei geni del procollageno (Col1 A1 e 2) variabilità del quadro clinico (anche 70 mutazioni) Le molecole anomale non formano la tripla elica
 A trasmissione autosomica dominante. Mutazioni in uno dei geni del procollageno (Col1 A1 e 2) variabilità del quadro clinico (anche 70 mutazioni) Le molecole anomale non formano la tripla elica.")
18
Osteogenesis-imperfecta
Gravità di quadri clinici variabili Bassa statura Fragilità ossea Sordità Denti opalescenti (al transilluminatore Deformazioni delle ossa lunghe Sclere di colore blu

19
Osteogenesis-imperfecta
Gravità di quadri clinici variabili Bassa statura Fragilità ossea Sordità Denti opalescenti (al transilluminatore) Altri tipi di OI, si verificano raramente, sono sottotipi della forma moderatamente grave (tipo IV). Le complicazioni sono connesse ai problemi con le ossa deboli e fratture multiple e possono comprendere: Perdita di udito (comune in tipo I e III); Insufficienza cardiaca (tipo II); Problemi respiratori e polmoniti a causa della deformità della parete toracica; Problemi al cavo spinale o tronco cerebrale; Permanente deformità.
 Altri tipi di OI, si verificano raramente, sono sottotipi della forma moderatamente grave (tipo IV). Le complicazioni sono connesse ai problemi con le ossa deboli e fratture multiple e possono comprendere: Perdita di udito (comune in tipo I e III); Insufficienza cardiaca (tipo II); Problemi respiratori e polmoniti a causa della deformità della parete toracica; Problemi al cavo spinale o tronco cerebrale; Permanente deformità.")
20
Osteogenesis imperfecta
Tipo I: ritardo di accrescimento nel 50%, fratture ossee; cifosi e scoliosi con iperestesibilità articolare, sclere bluastre e perdita dell'udito sia a difetto neurosensoriale che a causa di anomalie ossee dell'orecchio medio e interno. Talora si associa a dentinogenesi imperfecta. Tipo II: fatale durante la vita intrauterina o nel periodo perinatale. Fragilità ossea grave con fratture multiple che si manifestano nel feto in utero. Tipo III: fratture alla nascita con deformazioni progressive degli arti e cifoscoliosi, sclere normali, bassa statura, dentinogenesi imperfecta Tipo IV: la forma clinicamente meno grave, con statura normale o poco ridotta, fragilità ossea moderata, fratture postnatali, sclere normali, udito normale, deformità variabili. Talora si associa a dentinogenesi imperfecta La malattia colpisce maschi e femmine con rapporto 1:1 e con incidenza di 1/ nati vivi. L'osteogenesi imperfetta è una malattia genetica a trasmissione autosomica dominante per anomalie nella sintesi del collagene tipo I° per mutazione dei geni Col1A1 e 2. Crea problemi a carico dello scheletro, delle articolazioni, degli occhi, delle orecchie, della cute e dei denti. I fenotipi più gravi o letali sono la conseguenza di difetti genetici, che determinano molecole anomale di collagene che non riescono a formare la tripla elica. Clinicamente manifesta fragilità ossea ed è conosciuta anche come Malattia di Lobstein (tipo I°), Sindrome di Vrolik (tipo II°), Sindrome di van der Hoeve, Sindrome di Eddowes. Attualmente se ne conoscono sette tipologie varianti a diversa gravità, i quattro tipi storicamente noti sono: Tipo I: ritardo di accrescimento nel 50%, fratture ossee; cifosi e scoliosi con iperestesibilità articolare, sclere bluastre e perdita dell'udito sia a difetto neurosensoriale che a causa di anomalie ossee dell'orecchio medio e interno. In certi casi si associa a dentinogenesi imperfetta. Tipo II: è costantemente fatale durante la vita intrauterina o nel periodo perinatale. Accentuatissima fragilità ossea con fratture multiple che si manifestano quando il feto è ancora in utero. Tipo III: fratture alla nascita con deformazioni progressive degli arti e cifoscoliosi, sclere normali, bassa statura, dentinogenesi imperfetta comune. Tipo IV: la forma clinicamente meno grave, con statura normale o poco ridotta, fragilità ossea lieve o moderata, fratture postnatali, sclere normali, udito normale, deformità variabili. In certi casi si associa a dentinogenesi imperfetta. La malattia colpisce maschi e femmine con rapporto 1:1 e con incidenza di 1/ nati vivi.
, Sindrome di Vrolik (tipo II°), Sindrome di van der Hoeve, Sindrome di Eddowes. Attualmente se ne conoscono sette tipologie varianti a diversa gravità, i quattro tipi storicamente noti sono: Tipo I: ritardo di accrescimento nel 50%, fratture ossee; cifosi e scoliosi con iperestesibilità articolare, sclere bluastre e perdita dell udito sia a difetto neurosensoriale che a causa di anomalie ossee dell orecchio medio e interno. In certi casi si associa a dentinogenesi imperfetta. Tipo II: è costantemente fatale durante la vita intrauterina o nel periodo perinatale. Accentuatissima fragilità ossea con fratture multiple che si manifestano quando il feto è ancora in utero. Tipo III: fratture alla nascita con deformazioni progressive degli arti e cifoscoliosi, sclere normali, bassa statura, dentinogenesi imperfetta comune. Tipo IV: la forma clinicamente meno grave, con statura normale o poco ridotta, fragilità ossea lieve o moderata, fratture postnatali, sclere normali, udito normale, deformità variabili. In certi casi si associa a dentinogenesi imperfetta. La malattia colpisce maschi e femmine con rapporto 1:1 e con incidenza di 1/ nati vivi.")
21
Osteogenesis imperfecta Malati Famosi
Michel Petrucciani Pianista Jazz Michael J Anderson Attore Nabil Shaban Attore He Pingping Guinness World Records

22
Sindrome di Ehler-Danlos
comprende un gruppo clinicamente e geneticamente eterogeneo di patologie a carico del tessuto connettivo 11 varianti Lassità delle articolazioni Ematomi cutanei Cute sottile ed iper-estensibile Problemi agli organi interni La sindrome ha peculiarità particolarmente eterogenee e non solo ciascun tipo, ma anche ciascun paziente, rappresenta spesso una patologia a sé stante per espressività, tipologia e gravità dei sintomi. Lesioni gravi: Rottura dell’aorta e di altri organi cavi

23
Sindrome di Ehler-Danlos
Difetto molecolare riguarda gli enzimi necessari per le modificazioni post-traduzionali dei collageni di tipo I e III Classificazione e Tipi di EDS Classica (ex I e II) Ipermobile (ex III) Vascolare (ex IV) Cifoscoliotica (ex VI) Artrocalasia (ex VII a, VII b) Dermatosparassi (ex VII c) N.B.: La differenziazione tra le forme non è sempre netta e definita, infatti possono essere presenti sintomi sovrapposti tra le varie forme. forme più comuni forma più grave
 Ipermobile (ex III) Vascolare (ex IV) Cifoscoliotica (ex VI) Artrocalasia (ex VII a, VII b) Dermatosparassi (ex VII c) N.B.: La differenziazione tra le forme non è sempre netta e definita, infatti possono essere presenti sintomi sovrapposti tra le varie forme. forme più comuni. forma più grave.")
24
Sindrome di Ehler-Danlos
Tipo Classico I principali sintomi sono: Cute iperestensibile, vellutata, traslucida e sottile. Cicatrici estese e caratteristiche (a "carta di sigaretta"). Pseudotumori molluscoidi. Ipermobilità e lassità articolare. Lussazioni ed instabilità articolare. Scarso tono muscolare. Comparsa di lividi a seguito di traumi anche minimi. Ereditarietà di tipo autosomico dominante per un difetto nei geni COLA541, COLA5A2.
. Pseudotumori molluscoidi. Ipermobilità e lassità articolare. Lussazioni ed instabilità articolare. Scarso tono muscolare. Comparsa di lividi a seguito di traumi anche minimi. Ereditarietà di tipo autosomico. dominante per un difetto nei geni COLA541, COLA5A2.")
25
Sindrome di Ehler-Danlos
Tipo Ipermobile I principali sintomi sono: Lussazioni ed instabilità articolare accentuata. Dolore cronico, diffuso alle maggior parte delle articolazioni e frequenti lussazioni, anche per traumi di minima intensità. Scarso trofismo muscolare. Iperstensività della cute. Possono presentarsi anche disturbi cardiaci quale il prolasso della valvola mitralica. Ha un' ereditarietà di tipo autosomico dominante, la causa di questa forma non è ancora conosciuta.

26
Sindrome di Ehler-Danlos
Tipo Vascolare I principali sintomi sono: Rottura spontanea o eccessiva fragilità delle pareti delle arterie, che risultano ben visibili attraverso la cute traslucida. Facilità di ematomi, anche per minimi traumi. Cute sottile ed iperestensibile. Ipermobilità articolare (solitamente lieve) e volto caratteristico. Esoftalmo. Possibile rottura di organi interni molto irrorati (es: utero). Pneumotoraci spontanei. E' la forma più grave: la rottura spontanea di un'arteria o di un organo interno può causare il decesso del paziente per emorragia. Ereditarietà di tipo autosomico dominante. Difetto nel gene COL3A1.
 e volto caratteristico. Esoftalmo. Possibile rottura di organi interni molto irrorati (es: utero). Pneumotoraci spontanei. E la forma più grave: la rottura spontanea di un arteria o di un organo interno può causare il decesso del paziente per emorragia. Ereditarietà di tipo autosomico dominante. Difetto nel gene COL3A1.")
27
Sindrome di Ehler-Danlos
Tipo Cifoscoliotico I principali sintomi sono: Grave scoliosi congenita progressiva. Fragilità della sclera oculare. Lassità delle articolazioni. Ipotonia muscolare. Perdita della capacità di deambulazione. Lividi. Fragilità tissutale Ha un' ereditarietà di tipo autosomico recessivo dovuta ad un deficit dell'enzima lisil-ossidasi (o protein-lisina 6-ossidasi) coinvolto nelle modifiche post-traduzionali della lisina, nelle procatene alfa del collagene.), modificazione del gene PLOD1
 coinvolto nelle modifiche post-traduzionali della lisina, nelle procatene alfa del collagene.), modificazione del gene PLOD1.")
28
Sindrome di Ehler-Danlos
Tipo Artrocalasia I principali sintomi sono: Grave lassità delle articolazioni. Lussazioni frequenti. Ipotonia muscolare. Scoliosi. Bassa statura Ha un' ereditarietà ti tipo autosomico dominante ed è dovuta ad un difetto nel gene COL1A1, COL1A2.

29
Sindrome di Ehler-Danlos
Tipo Dermatosparassi I principali sintomi sono: Grave fragilità cutanea. Cute iperestensibile. Faclie formazione di ecchimosi e lividi. Ha un' ereditarietà di tipo autosomico recessivo ed è dovuta alla mancanza del procollagene 1 N-peptidase per alterazione del gene ADAMST2

30
Altri Difetti Genetici
Forme latenti, frequenti Prolasso valvolare mitralico Aneurismi aortici Osteoartrite Certe forme di osteoporosi

31
Fibre collagene difettose
Difetti acquisiti Carenze Nutrizionali: Deficiente idrossilazione da carenza di Vit. C (scorbuto) Collageno con emivita breve (cadute dentali, sanguinamenti, deiscenza delle cicatrici) < Idrossilazione - Cross-linking Fibre collagene difettose
 Collageno con emivita breve (cadute dentali, sanguinamenti, deiscenza delle cicatrici) < Idrossilazione. - Cross-linking. Fibre collagene difettose.")
32
Difetti di cross-linking
Diminuito cross-linking Omocistinuria: malattia genetica autosomica recessiva per mutazione di un gene sul braccio lungo del cromosoma 21. Manifestazioni cliniche: un fenotipo "marfanoide", con corporatura alta e sottile, aracnodattilia, pectus excavatum, cifoscoliosi ed osteoporosi; Danni al cristallino con miopia per lussazione del cristallino Ritardo mentale (60% dei pazienti) e comportamenti schizofrenici. Danni cardiovascolari: trombosi arteriose e venose, degenerazione dell'aorta e delle grandi arterie, iperplasia e fibrosi dello strato intimale dei vasi arteriosi e venosi. trombosi venosa profonda complicata spesso da embolia polmonare e trombosi arteriosa a livello coronarico, cerebrale e periferico. l’omocisteina blocca la formazione di legami crociati stabili Eccessivo cross-linking: invecchiamento e diabete
 e comportamenti schizofrenici. Danni cardiovascolari: trombosi arteriose e venose, degenerazione dell aorta e delle grandi arterie, iperplasia e fibrosi dello strato intimale dei vasi arteriosi e venosi. trombosi venosa profonda complicata spesso da embolia polmonare e trombosi arteriosa a livello coronarico, cerebrale e periferico. l’omocisteina blocca la formazione di legami crociati stabili. Eccessivo cross-linking: invecchiamento e diabete.")
33
Difetti di cross-linking
Eccessivo cross-linking: invecchiamento e diabete

34
Digestione ad opera di enzimi
Normalmente le collagenasi sono sotto il controllo di potenti inibitori Intervengono nel catabolismo nel ricambio del collageno: Guarigione delle Ferite Riassorbimento dell’osso Patologie: Collagenasi prodotte da fibroblasti, cellule infiammatorie, batteri Osteoartrite o artrosi distruzione della matrice cartilaginea ialina Malattie della cornea Collagenasi batteriche. Es: Clostridium hystoliticum (cangrena gassosa)
")
35
Altre Patologie Calcificazione del collageno: invecchiamento tendini
Alcaptonuria (ocronosi) per carenza di omogentisinico –ossidasi accumulo di ac. omogentisinico che denatura il collageno artrite cronica progressiva Malattia ereditaria. La carenza dell'enzima omogentisinico ossidasi, bloccando la demolizione di fenilalanina e tirosina determina l'accumulo di pigmenti detti alcaptoni. I segni caratteristici sono: Aspetto bruno-azzurro della pelle, evidente sulla punta del naso, alle orecchie, alle sclere. Artropatie diffuse. Presenza degli alcaptoni nelle urine (di colore scuro e diventano nere se esposte alla luce (si può riscontrare nei pannolini dei lattanti). L'alcaptonuria non incide sulla durata media della vita; l'interessamento delle articolazioni è l'aspetto più grave e invalidante.
 per carenza di omogentisinico –ossidasi accumulo di ac. omogentisinico che denatura il collageno artrite cronica progressiva. Malattia ereditaria. La carenza dell enzima omogentisinico ossidasi, bloccando la demolizione di fenilalanina e tirosina determina l accumulo di pigmenti detti alcaptoni. I segni caratteristici sono: Aspetto bruno-azzurro della pelle, evidente sulla punta del naso, alle orecchie, alle sclere. Artropatie diffuse. Presenza degli alcaptoni nelle urine (di colore scuro. e diventano nere se esposte alla luce (si può riscontrare. nei pannolini dei lattanti). L alcaptonuria non incide sulla durata media della vita; l interessamento delle articolazioni è l aspetto più grave e invalidante.")
36
Altre Patologie Epidermolisi bollosa: mancanza di fibrille di ancoraggio- filamenti di collageno di tipo VII- tra membrana basale e derma: 10-14 malattie genetiche della cute e delle mucose caratterizzate da sviluppo di bolle in seguito a traumi anche minimi Cicatrici interdigitali Contrattura delle articolazioni Cicatrici corneali Infezioni croniche

37
Elastina E’ prodotta da: fibroblasti e cellule muscolari lisce

38
Elastina Condivide con il collageno il cross-linking e l’enzima responsabile lisil-ossidasi Le catene polipeptidiche di elastina formano fibre elastiche estensibili. La differenza è dovuta alle differenti sequenze aminoacidiche (2 lisina + 2 lisina desmosina ) Il collageno è una tripla elica, formata da tre catene proteiche. Nello spazio extracellulare formano fibre inestensibili
 Il collageno è una tripla elica, formata da tre catene proteiche. Nello spazio extracellulare formano fibre inestensibili.")
39
Patologia dell’elastina Difetti genetici
Cutis laxa -> riduzione di fibre elastiche

40
Sindrome di Marfan E’ una malattia autosomica dominante legata al gene FBN1 sul cromosoma 15, che codifica per una proteina la fibrillina, essenziale per la formazione delle fibre elastiche. Senza il supporto della fibrillina molti tessuti sono deboli (rottura dei vasi) Le manifestazioni sono a carico dello: Scheletro (deformità della colonna vertebrale) Ipermobilità articolare Polmoni Cuore e vasi sanguigni (prolasso mitralico) Caratterizzata da arti insolitamente lunghi (aracnodattilia) Disturbi oculari
 Le manifestazioni sono a carico dello: Scheletro (deformità della colonna vertebrale) Ipermobilità articolare. Polmoni. Cuore e vasi sanguigni (prolasso mitralico) Caratterizzata da arti insolitamente. lunghi (aracnodattilia) Disturbi oculari.")
41
Difetti acquisiti Elastosi solare o dermatoeliosi o dermatosi attinica
Invecchiamento Digestione ad opera di elastasi Enfisema Veleni di serpenti Batteri (clostridium hystoliticum)
")
42
Digestione ad opera di elastasi
Enfisema Veleni di serpenti Batteri (clostridium hystoliticum)
")
43
Membrane basali Sono strati sottili di matrice extracellulare specializzata che separano le cellule che la sintetizzano dal tessuto connettivo: Endotelio capillare Cell. Muscolari Epitelio Adipociti Cell.di Schwann

45
Membrane basali . A current model of the molecular structure of a basal lamina. The basal lamina (A) is formed by specific interactions between the proteins type IV collagen, laminin, and entactin plus the proteoglycan perlecan (B). Arrows in (B) connect molecules that can bind directly to each other.
 is formed by specific interactions between the proteins type IV collagen, laminin, and entactin plus the proteoglycan perlecan (B). Arrows in (B) connect molecules that can bind directly to each other.")
46
Ispessimento patologico
Riflette uno stato irritativo delle cellule che producono la membrana basale Mucosa bronchiale dei soggetti asmaticilamina vitrea Soggetti diabetici (mb pericapillari) si formano prodotti della glicosilazione avanzata (AGE Advanced glycosylation end-products) a carico delle macromolecole della MB Soggetti affetti da porfiria Eccessive assunzioni di Fenacitina ispessimento dei tessuti renali e pararenali
 si formano prodotti della glicosilazione avanzata (AGE Advanced glycosylation end-products) a carico delle macromolecole della MB. Soggetti affetti da porfiria. Eccessive assunzioni di Fenacitina ispessimento dei tessuti renali e pararenali.")
47
Proteoglicani Componenti di: Interstizio connettivale Membrane basali
Membrane cellulari Coinvolti nel: Riconoscimento cellulare Adesione Crescita Mantengono liquidi nel Comp. Interstiziale Proteggono le cartilagini dal carico meccanico Sintetizzati da condrociti

48
Patologia dei proteoglicani
Patologia delle articolazioni Degenerazione extracellulare della cartilagine: Artrite degenerativa o artrosi Riduzione di proteoglicani Mixedema o edema mucoso: è dovuto all’accumulo di proteoglicani Ipotiroidismo avanzato (accumulo di ac. Ialuronico nell’interstizio dei tessuti) Ma anche nella malattia di Graves (mixedema pretibiale)
 Ma anche nella malattia di Graves (mixedema pretibiale)")
Presentazioni simili