Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
CITOGENETICA CONVENZIONALE E MOLECOLARE

2
CITOGENETICA CONVENZIONALE
La citogenetica convenzionale è una tecnica che permette lo studio del numero e della struttura dei cromosomi (studio del cariotipo) I cromosomi vengono esaminati “bloccati” in metafase. Step: prelievo del campione (SP, BM o tessuto linf.) allestimento delle colture cellulari allestimento dei preparati bandeggio dei cromosomi
 I cromosomi vengono esaminati bloccati in. metafase. Step: prelievo del campione (SP, BM o tessuto linf.) allestimento delle colture cellulari. allestimento dei preparati. bandeggio dei cromosomi.")
3
Allestimento delle colture e dei preparati
Diretta: terreno di coltura+colchicina 4-6 ore Medio termine: terreno + colchicina 24-48 ore Sincronizzata: blocco in fase S (MTX) rimozione del blocco colchicina Tecniche di coltura post-incubazione: centrifugazione soluzione ipotonica per distensione dei cromosomi centrifugazione → fissativo (alcol metilico o acido acetico) Allestimento preparati
 rimozione del blocco. colchicina. Tecniche di. coltura. post-incubazione: centrifugazione. soluzione ipotonica per distensione. dei cromosomi. centrifugazione → fissativo (alcol metilico o. acido acetico) Allestimento. preparati.")
4
Tecniche di bandeggio dei cromosomi
Bandeggio Q Bandeggio G Bandeggio R Tecniche generali (intero cromosoma) BANDEGGIO STATICO Bandeggio C Bandeggio Cd (fuso) Bandeggio NOR geni RNA ribosomi Tecniche speciali (porzioni cromosoma) Informazioni sul tipo di replica di ogni cromosoma durante la fase S (uso raro in onco-ematologia) BANDEGGIO DINAMICO
 BANDEGGIO. STATICO. Bandeggio C. Bandeggio Cd (fuso) Bandeggio NOR geni. RNA ribosomi. Tecniche speciali. (porzioni cromosoma) Informazioni sul tipo di replica di. ogni cromosoma durante la fase S. (uso raro in onco-ematologia) BANDEGGIO. DINAMICO.")
5
Bandeggio Q mostarda di chinacrina contrasto tra bande: scarso
(intercala tra copppie di basi) contrasto tra bande: scarso non colora telomeri colora eterocromatina
 contrasto tra. bande: scarso. non colora. telomeri. colora. eterocromatina.")
6
Bandeggio R soluzione salina + Giemsa complementari a bande Q e G
contrasto < a bandeggio G colora telomeri eucromatina colora poco eterocromatina

7
Cromosomi normali Cromosoma metacentrico Cromosoma submetacentrico
acrocentrico

8
Cariotipo normale 46 cromosomi (n. diploide) 22 coppie (omologhi)
autosomi (1-22) 2 cromosomi sessuali XY (M) e XX (F)
 2 cromosomi sessuali. XY (M) e XX (F)")
9
Anomalie cromosomiche
quasi aploidi (n +/-) ipodiploidi (2n-) iperdiploidi (2n+) pseudodiploidi monosomie trisomie Anomalie del numero traslocazioni (reciproche e non) delezioni (intersiziali o terminali) duplicazioni inversioni Anomalie strutturali
 ipodiploidi (2n-) iperdiploidi (2n+) pseudodiploidi. monosomie. trisomie. Anomalie del numero. traslocazioni (reciproche e non) delezioni (intersiziali o terminali) duplicazioni. inversioni. Anomalie strutturali.")
10
Traslocazioni T (2;15) (p11.2;q11.2) T (13;14) (p11.2;p11.2)
bilanciata sbilanciata

11
Delezioni cromosomiche
(q11.23 q21.2) delezione interstiziale braccio lungo (q) cromosoma 7
 delezione. interstiziale. braccio lungo (q) cromosoma 7.")
12
CITOGENETICA MOLECOLARE (FISH)
La Fluorescent In Situ Hybridization (FISH) è una tecnologia che utilizza sonde nucleotidiche marcate (DNA probes) per identificare specifiche regioni di un cromosoma ovvero determinate sequenze del DNA. Step: denaturazione del DNA incubazione sonda + DNA denaturato (“annealing”) rilevazione del segnale della sonda con microscopio a fluorescenza
 è una tecnologia che utilizza. sonde nucleotidiche marcate (DNA. probes) per identificare specifiche. regioni di un cromosoma ovvero. determinate sequenze del DNA. Step: denaturazione del DNA. incubazione sonda + DNA denaturato. ( annealing ) rilevazione del segnale della sonda. con microscopio a fluorescenza.")
13
Tipi di sonde (DNA probe)
Nucleotidi + biotina o digossigenina (fluorocromo + streptavidina) (fluorocromo + Ac anti-digossigenina) Sonde nucleotidi coniugati a fluorocromi Alfoidi: per sequenze ripetitive dei satelliti (anomalie numeriche) Painting: specifiche per un cromosoma (cromosomi molto riarrangiati) Locus singolo: sequenze specifiche DNA (es. geni di fusione) Sonde
 (fluorocromo + Ac anti-digossigenina) Sonde. nucleotidi coniugati a fluorocromi. Alfoidi: per sequenze ripetitive dei satelliti. (anomalie numeriche) Painting: specifiche per un cromosoma. (cromosomi molto riarrangiati) Locus singolo: sequenze specifiche DNA. (es. geni di fusione) Sonde.")
14
FISH: tappe della metodica
Campioni DNA: cromosomi in metafase o nuclei in interfase

15
FISH in interfase

16
FISH in metafase probe per parte terminale del cr. 4q

17
FISH: vantaggi e limiti
Esamina elevato n. di cellule in tempi brevi Metodica semplice Elevata efficienza di ibridizzazione Elevata sensibilità e specificità Non necessità cellule in mitosi Correla dato citogenetico con morfologico Vantaggi Informazioni su singolo cromosoma/gene Esame simultaneo di pochi DNA bersaglio Soglia di positività da calcolare per ogni sonda Possibili artefatti nell’analisi di inclusi in paraffina Limiti

18
Citogenetica nelle leucemie acute
Classificazione delle leucemie acute (entità cliniche all’interno di un citotipo FAB) Definizione del rischio citogenetico (significato prognostico) Monitoraggio della malattia minima residua: sensibilità citogenetica convenzionale < sensibilità FISH
 Definizione del rischio citogenetico. (significato prognostico) Monitoraggio della malattia minima residua: sensibilità citogenetica convenzionale < sensibilità FISH.")
19
TECNICHE DI BIOLOGIA MOLECOLARE

20
POLYMERASE CHAIN REACTION (PCR)
Tecnica che si basa sulla capacità di una DNA polimerasi di amplificare in modo esponenziale una regione di DNA a sequenza sconosciuta (templato) posta tra due porzioni di DNA a sequenza nota. Step: Estrazione di DNA (o RNA) Cicli di amplificazione: denaturazione “annealing” estensione dei primers Analisi degli amplificati
 posta tra due porzioni di. DNA a sequenza nota. Step: Estrazione di DNA (o RNA) Cicli di amplificazione: denaturazione. annealing estensione dei primers. Analisi degli amplificati.")
21
PCR: estrazione di DNA (o RNA)
Proteinasi + fenolo-cloroformio-alcool isoamilico time: 2 giorni DNA Guanidio isotiocianato + mercaptoetanolo cloroformio ( a 4 °C) time: 2 giorni RNA DNA/RNA KIT commerciali/ strumenti per automazione 260 nm (DNA) 280 nm (proteine) Concentrazione Qualità A 260 A 280 spettrofotometria
 time: 2 giorni. RNA. DNA/RNA. KIT commerciali/ strumenti per automazione. 260 nm (DNA) 280 nm (proteine) Concentrazione. Qualità. A 260. A 280. spettrofotometria.")
22
PCR: cicli di amplificazione
Miscela di amplificazione DNA templato DNA polimerasi termostabile Buffer di reazione (TrisHCl e KCl) Ione magnesio Desossinucleotidi trifosfato Primers: brevi sequenze di DNA a singolo complementari a sequenze note poste a monte e a valle del templato
 Ione magnesio. Desossinucleotidi trifosfato. Primers: brevi sequenze di DNA a singolo. complementari a sequenze note. poste a monte e a valle del templato.")
23
PCR: cicli di amplificazione
Denaturazione (~ 1 min 95°C) Raffreddamento e “annealing” dei primers (~ 1 min 45-60°C) Estensione dei primers da DNA polimerasi (~ 1 min 72°C)
 Raffreddamento e. annealing dei primers. (~ 1 min 45-60°C) Estensione dei primers. da DNA polimerasi. (~ 1 min 72°C)")
24
PCR: cicli di amplificazione
Cicli ripetuti volte

25
crescita esponenziale ed efficienza di reazione
Amplificazione: crescita esponenziale ed efficienza di reazione Xn = X0 x (1 + Ex)n Qualità e concentrazione del DNA/RNA Qualità e concentrazione del cDNA Concentrazione dei vari reagenti Condizioni di temperatura della reazione Numero di cicli plateau Efficienza della reazione
n. Qualità e concentrazione del DNA/RNA. Qualità e concentrazione del cDNA. Concentrazione dei vari reagenti. Condizioni di temperatura della reazione. Numero di cicli plateau. Efficienza della. reazione.")
26
PCR qualitativa: analisi dei risultati
Elettroforesi in gel di agarosio Corsa: 1/2 ora a 150 V Colorazione con etidio bromuro UV transilluminazione Fotografia

27
Fig. 4 Roccaro et al. VEGF IL-6 GAPDH GAPDH Ang1 Ang2 IGF-1 GAPDH
ddH2O 0 nM 5 nM 7.5 nM ddH2O 0 nM 5 nM 7.5 nM VEGF IL-6 GAPDH GAPDH 150 150 120 Interior area (pixel) 100 Interior area (pixel) 90 50 60 30 neg ctrl Bortezomib [nM] 5 7.5 neg ctrl Bortezomib [nM] 5 7.5 ddH2O 0 nM 5 nM 7.5 nM ddH2O 0 nM 5 nM 7.5 nM ddH2O 0 nM 5 nM 7.5 nM Ang1 Ang2 IGF-1 GAPDH GAPDH GAPDH 100 150 80 180 100 Interior area (pixel) 60 Interior area (pixel) 40 50 Interior area (pixel) 120 20 60 neg ctrl Bortezomib [nM] 5 7.5 neg ctrl Bortezomib [nM] 5 7.5 neg ctrl Bortezomib [nM] 5 7.5 Fig. 4 Roccaro et al.
 100. Interior area (pixel) neg ctrl. Bortezomib [nM] neg ctrl. Bortezomib [nM] ddH2O. 0 nM. 5 nM. 7.5 nM. ddH2O. 0 nM. 5 nM. 7.5 nM. ddH2O. 0 nM. 5 nM. 7.5 nM. Ang1. Ang2. IGF-1. GAPDH. GAPDH. GAPDH Interior area (pixel) 60. Interior area (pixel) Interior area (pixel) neg ctrl. Bortezomib [nM] neg ctrl. Bortezomib [nM] neg ctrl. Bortezomib [nM] Fig. 4. Roccaro et al.")
28
PCR qualitativa: vantaggi e limiti
Elevata sensibilità Elevata specificità Identificazione di traslocazioni non dimostrate dalla citogenetica Possibilità di analisi simultanea delle traslocazioni più frequenti Possibilità dell’esecuzione dell’analisi da campioni bioptici Vantaggi Possibilità di contaminazioni Efficienza di amplificazione variabile e dipendente da vari fattori Impossibilità di stabilire la q di sequenza bersaglio Presente all’inizio nel campione Limiti

29
PCR quantitativa real-time
Thermal-cycler (amplificazione) Sistema ottico per rilievo della fluorescenza Software per raccolta ed analisi dei dati Analisi dei prodotti non alla fine della reazione, ma durante la fase di crescita lineare delle molecole di amplificato
 Sistema ottico per rilievo della. fluorescenza. Software per raccolta ed analisi. dei dati. Analisi dei prodotti non alla fine. della reazione, ma durante la. fase di crescita lineare delle. molecole di amplificato.")
30
Real-time PCR: metodo Taqman
Sonda specifica per target Q = fluorocromo quencher (rosso, onda lunga) R = fluorocromo reporter (verde, onda corta) Sonda Taqman si lega a DNA target I primer si legano a 3’ e 5’ del DNA templato
 R = fluorocromo reporter. (verde, onda corta) Sonda Taqman si lega a. DNA target. I primer si legano a 3’ e 5’ del DNA templato.")
31
Real-time PCR: metodo Taqman

32
Real-time PCR: metodo Taqman

33
Real-time PCR: metodo Taqman

34
PCR quantitativa: vantaggi e limiti
Velocità Sensibilità (10-4 – 10-6) Specificità Standards commutabilità confrontabilità accuratezza Vantaggi Variabilità nella determinazione quantitativa Complessità sperimentale Mancanza di standards Valutazione critica dei risultati Limiti
 Specificità. Standards commutabilità. confrontabilità. accuratezza. Vantaggi. Variabilità nella determinazione quantitativa. Complessità sperimentale. Mancanza di standards. Valutazione critica dei risultati. Limiti.")
35
TECNOLOGIA DEI MICROARRAY E LEUCEMIE ACUTE

36
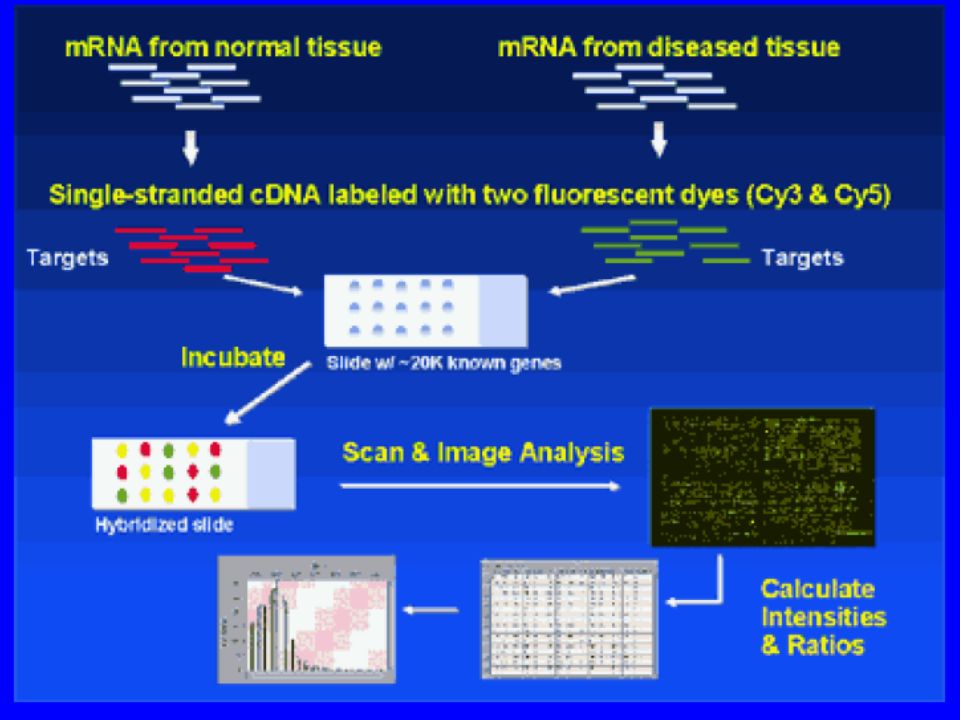
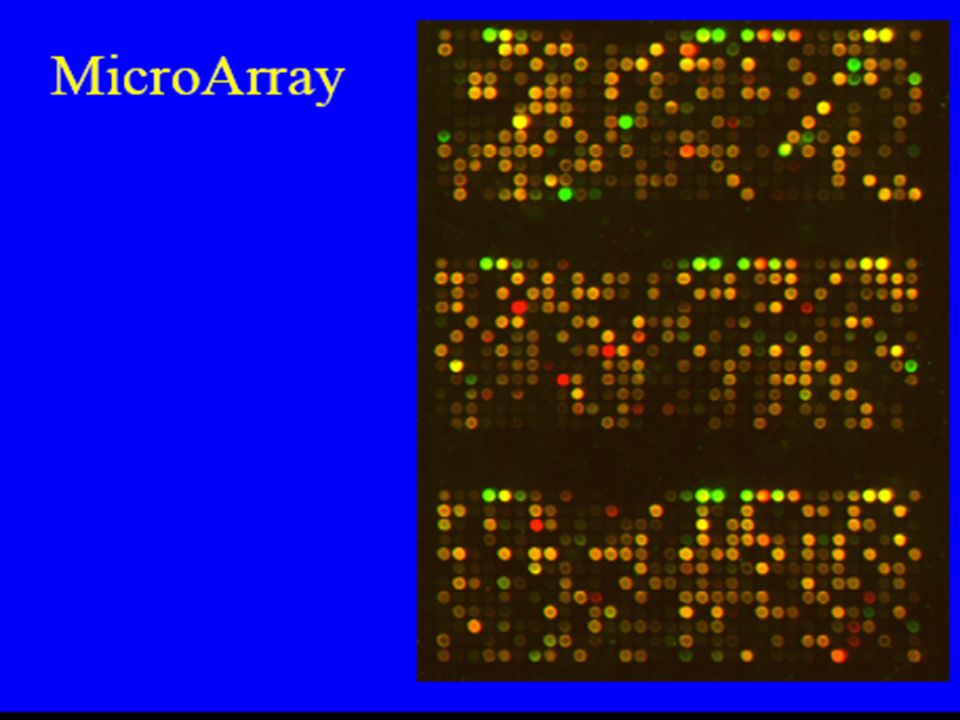
DNA-microarray Metodica che consente di analizzare il profilo di espressione genica delle cellule. Procedura di base: Sequenze di DNA sono schierate in ordine su supporto solido. Dal campione viene estratto mRNA e retrotrascritto in cDNA. cDNA, marcato con tracciante fluorescente, ibridizza con sequenze su array. Il livello di espressione di un gene è direttamente proporzionale all’intensità di segnale derivato dalle immagini digitali acquisite. Principali tipi di microarray: 1. membrane based cDNA microarray 2. glass slide-based cDNA microarrays 3. oligonucleotide microarray o DNA chip

40
DNA-microarray e leucemie acute: possibili sviluppi
Riclassificare le leucemie Individuare profili di espressione genica in relazione alla prognosi Individuare profili di espressione genica in relazione a vie di trasduzione del segnale per definire terapie molecolari target Individuare profili di espressione genica in relazione alla sensibilità o resistenza ad un dato farmaco

Presentazioni simili

















>")


