Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
MALATTIE INTERSTIZIALI POLMONARI
Definizione Le malattie interstiziali del polmone, o malattie polmonari parenchimali diffuse, sono processi patologici caratterizzati da distinti infiltrati cellulari e deposizione di matrice extracellulare (proteine, collagene) nelle regioni polmonari a valle del bronchiolo terminale (acino). Sebbene i processi infettivi polmonari e alcune forme neoplastiche possono interessare in maniera diffusa le regioni acinari del polmone, tali patologie sono generalmente non considerate come malattie interstiziali diffuse ma devono essere prese in considerazione nella diagnostica differenziale.
 nelle regioni polmonari a valle del bronchiolo terminale (acino). Sebbene i processi infettivi polmonari e alcune forme neoplastiche possono interessare in maniera diffusa le regioni acinari del polmone, tali patologie sono generalmente non considerate come malattie interstiziali diffuse ma devono essere prese in considerazione nella diagnostica differenziale.")
2
Da cause ambientali (pneumoconiosi)
Classificazione (1°) Potrebbero essere classificate in: ad etiologia conosciuta o sconosciuta. Da cause ambientali (pneumoconiosi) - organiche (alveoliti allergiche strinseche) - inorganiche fibrosanti (silicosi, asbestosi, berilliosi, metalli pesanti, ecc) - non fibrosanti o da accumulo (siderosi, ecc) Da farmaci Chemoterapici, cardiovascolari, radiazioni, pesticidi, ecc Malattie granulomatose Sarcoidosi, berilliosi, AAE, Wegener, Churg-Strauss, Istiocitosi a cellule di Langherans, ecc.
 Potrebbero essere classificate in: ad etiologia conosciuta o sconosciuta. Da cause ambientali (pneumoconiosi) - organiche (alveoliti allergiche strinseche) - inorganiche fibrosanti (silicosi, asbestosi, berilliosi, metalli pesanti, ecc) - non fibrosanti o da accumulo (siderosi, ecc) Da farmaci. Chemoterapici, cardiovascolari, radiazioni, pesticidi, ecc. Malattie granulomatose. Sarcoidosi, berilliosi, AAE, Wegener, Churg-Strauss, Istiocitosi a cellule di Langherans, ecc.")
3
Polmoniti Interstiziali Idiopatiche
Classificazione (2°) In corso di malattie sistemiche - Connettiviti (SSP, RA, LES, ecc) - Vasculiti (Churg-Strauss, Wegener, arterite di Takayasu e a cellule giganti, Sindr. Di Good-Pasture, ecc). - Vascolari (Sindr. da Ac antifosfolipidi, Ipertensione Polmonare Primitiva, Emosiderosi polmonare idiopatica, ecc). Polmoniti Interstiziali Idiopatiche - Fibrosi Polmonare Idiopatica (IPF) - Altre polmoniti interstiziali oltre la IPF (NSIP, DIP,BOOP, ecc). Altre malattie diffuse polmonari Linfangioleiomiomatosi (LAM), Proteinosi alveolare, microlitiasi alveolare, amiloidosi, scompenso cardiaco congestizio cronico, LIP, ecc).
 In corso di malattie sistemiche. - Connettiviti (SSP, RA, LES, ecc) - Vasculiti (Churg-Strauss, Wegener, arterite di Takayasu e a cellule giganti, Sindr. Di Good-Pasture, ecc). - Vascolari (Sindr. da Ac antifosfolipidi, Ipertensione Polmonare Primitiva, Emosiderosi polmonare idiopatica, ecc). Polmoniti Interstiziali Idiopatiche. - Fibrosi Polmonare Idiopatica (IPF) - Altre polmoniti interstiziali oltre la IPF (NSIP, DIP,BOOP, ecc). Altre malattie diffuse polmonari. Linfangioleiomiomatosi (LAM), Proteinosi alveolare, microlitiasi alveolare, amiloidosi, scompenso cardiaco congestizio cronico, LIP, ecc).")
4
APPROCCIO DIAGNOSTICO
1a fase - Anamnesi - Esame obiettivo - Rx del torace - Prove di funzionalità ventilatoria - Esami ematochimici selettivi 2a fase - HRTC - BAL - Biopsia polmonare

5
- anamnesi postiva per malattie del connettivo o vasculiti
Fisiologica - ambientale domestica (vicinanza a fabbriche/lavorazioni, hobby, animali domestici, ecc) - ambientale lavorativa (AAE, polveri organiche e inorganiche, ecc) - viaggi (possibilità di infestazioni parassitiche con susseguente eosinofilia polmonare) - tabagismo - assunzione di farmaci (amiodarone, chemioterapici, radiazioni, ecc (*) Patologica remota - anamnesi postiva per malattie del connettivo o vasculiti - patologie che hanno richiesto/richiedono trattamenti farmacologici a lungo termine - malattie neoplastiche con trattamenti radianti e/o chemioterapici - malattie neoplastiche che possono determinare linfangite carcinomatosa (*)
 - ambientale lavorativa (AAE, polveri organiche e inorganiche, ecc) - viaggi (possibilità di infestazioni parassitiche con susseguente eosinofilia polmonare) - tabagismo. - assunzione di farmaci (amiodarone, chemioterapici, radiazioni, ecc (*) Patologica remota. - anamnesi postiva per malattie del connettivo o vasculiti. - patologie che hanno richiesto/richiedono trattamenti farmacologici a lungo termine. - malattie neoplastiche con trattamenti radianti e/o chemioterapici. - malattie neoplastiche che possono determinare linfangite carcinomatosa. (*)")
6
Patologica prossima (modalità di comparsa di sintomi e segni)
ANAMNESI Patologica prossima (modalità di comparsa di sintomi e segni) Dispnea progressiva Tosse con/senza espettorazione (generalmente secca in fase iniziale) Rapidità d’insorgenza dei sintomi (acuta in: ARDS, infezioni, edema polmonare, AAE, emorragia alveolare diffusa, ecc.) Età di comparsa dei sintomi: aa (sarcoidosi, linfangioleiomiomatosi, istiocitosi, ecc) - >50 aa (IPF o UIP, ecc) Eventuale pleurite (malattie del connettivo) Eventuale emottisi (emorragia alveolare) Eventuale pneumotorace (linfangioleiomiomatosi, istiocitosi a cellule di Langherans) ESAME OBIETTIVO Paucisintomatico. Possibile ippocratismo digitale e crepitii bibasali (fine-inspirazione) (crackles) nella IPF. Possibili segni a carico di altri organi e apparati (connettiviti, sarcoidosi, ecc)
 Dispnea progressiva. Tosse con/senza espettorazione (generalmente secca in fase iniziale) Rapidità d’insorgenza dei sintomi (acuta in: ARDS, infezioni, edema polmonare, AAE, emorragia alveolare diffusa, ecc.) Età di comparsa dei sintomi: aa (sarcoidosi, linfangioleiomiomatosi, istiocitosi, ecc) - >50 aa (IPF o UIP, ecc) Eventuale pleurite (malattie del connettivo) Eventuale emottisi (emorragia alveolare) Eventuale pneumotorace (linfangioleiomiomatosi, istiocitosi a cellule di Langherans) ESAME OBIETTIVO. Paucisintomatico. Possibile ippocratismo digitale e crepitii bibasali (fine-inspirazione) (crackles) nella IPF. Possibili segni a carico di altri organi e apparati (connettiviti, sarcoidosi, ecc)")
7
RADIOGRAFIA DEL TORACE
Dimensione e forma delle anormalità - puntiformi (= 1 mm): microlitiasi alveolare, tubercolosi miliare, emosiderosi polmonare idiopatica. - nodulari (< 5 mm): silicosi, AEE, sarcoidosi. - macronodulari (> 5 mm): AR, granulomatosi di Wegener (escavati), linfomi, altre neoplasie. - reticolo-nodulari: asbestosi. - lineari settali (strie di Kerley): scompenso cardiaco congestizio, LAM
: microlitiasi alveolare, tubercolosi miliare, emosiderosi polmonare idiopatica. - nodulari (< 5 mm): silicosi, AEE, sarcoidosi. - macronodulari (> 5 mm): AR, granulomatosi di Wegener (escavati), linfomi, altre neoplasie. - reticolo-nodulari: asbestosi. - lineari settali (strie di Kerley): scompenso cardiaco congestizio, LAM.")
8
MICROLITIASI ALVEOLARE

9
MICROLITIASI ALVEOLARE

10
SILICOSI

11
AR

12
LINFANGIOLEIOMIOMATOSI

13
LINFANGIOLEIOMIOMATOSI

14
RADIOGRAFIA DEL TORACE
Dimensione e forma delle anormalità - puntiformi (= 1 mm): microlitiasi alveolare, tubercolosi miliare, emosiderosi polmonare idiopatica. - nodulari (< 5 mm): silicosi, AEE, sarcoidosi. - macronodulari (> 5 mm): AR, granulomatosi di Wegener (escavati), linfomi, altre neoplasie. - reticolo-nodulari: asbestosi. - lineari settali (strie di Kerley): scompenso cardiaco congestizio, LAM Distribuzione delle anormalità - centrale o periferica - apici: sarcoidosi e tbc, silicosi - medio-toraciche: sarcoidosi e AAE croniche - basi: IPF, asbestosi (reticolari o reticolonodulari, periferiche, oscuranti i contorni cardiaci e del diaframma).
: microlitiasi alveolare, tubercolosi miliare, emosiderosi polmonare idiopatica. - nodulari (< 5 mm): silicosi, AEE, sarcoidosi. - macronodulari (> 5 mm): AR, granulomatosi di Wegener (escavati), linfomi, altre neoplasie. - reticolo-nodulari: asbestosi. - lineari settali (strie di Kerley): scompenso cardiaco congestizio, LAM. Distribuzione delle anormalità. - centrale o periferica. - apici: sarcoidosi e tbc, silicosi. - medio-toraciche: sarcoidosi e AAE croniche. - basi: IPF, asbestosi (reticolari o reticolonodulari, periferiche, oscuranti i contorni cardiaci e del diaframma).")
15
FIBROSI POLMONARE IDIOPATICA
Reticolo-micronodulazione periferica prevalente alle regioni basali oscurante i contorni diaframmatici e cardiaci

16
RADIOGRAFIA DEL TORACE
Confluenza La confluenza delle opacità denota il riempimento degli spazi aerei (alveoli). La presenza di un broncogramma aereo è molto utile nel definire un consolidamento polmonare (proteinosi alveolare, emorragia alveolare diffusa, carcinoma bronchiolo-alveolare, linfoma, ecc).
. La presenza di un broncogramma aereo è molto utile nel definire un consolidamento polmonare (proteinosi alveolare, emorragia alveolare diffusa, carcinoma bronchiolo-alveolare, linfoma, ecc).")
17
PROTEINOSI POLMONARE ALVEOLARE

18
PROTEINOSI ALVEOLARE

19
RADIOGRAFIA DEL TORACE
Confluenza La confluenza delle opacità denota il riempimento degli spazi aerei (alveoli). La presenza di un broncogramma aereo è molto utile nel definire un consolidamento polmonare (proteinosi alveolare, emorragia alveolare diffusa, carcinoma bronchiolo-alveolare, linfoma, ecc). Coinvolgimento pleurico e linfoadenopatie mediastiniche Il coinvolgimento pleurico (con/senza versamento) è caratteristico dell’asbestosi (placche pleuriche), delle connettiviti (versamento pleurico in AR e LES), chilotorace (LAM). La presenza di adenopatie ilari, spesso simmetriche, è caratteristico della sarcoidosi; può essere altresì presente, generalmente monolaterali, nella tbc e nei linfomi. La contemporanea presenza di adenopatie calcifiche spesso si riscontrano nella silicosi e nella tbc (talvolta nella sarcoidosi cronica).
. La presenza di un broncogramma aereo è molto utile nel definire un consolidamento polmonare (proteinosi alveolare, emorragia alveolare diffusa, carcinoma bronchiolo-alveolare, linfoma, ecc). Coinvolgimento pleurico e linfoadenopatie mediastiniche. Il coinvolgimento pleurico (con/senza versamento) è caratteristico dell’asbestosi (placche pleuriche), delle connettiviti (versamento pleurico in AR e LES), chilotorace (LAM). La presenza di adenopatie ilari, spesso simmetriche, è caratteristico della sarcoidosi; può essere altresì presente, generalmente monolaterali, nella tbc e nei linfomi. La contemporanea presenza di adenopatie calcifiche spesso si riscontrano nella silicosi e nella tbc (talvolta nella sarcoidosi cronica).")
20
ASBESTOSI

21
ASBESTOSI

23
Prove di funzionalita’ ventilatoria : nella maggioranza dei pazienti con pneumopatia diffusa è presente un deficit ventilatorio di tipo restrittivo. In pneumopatie diffuse in fase avanzata e nelle quali una compromissione delle vie aeree è associata al danno parenchimale, può essere presente un deficit ventilatorio di tipo misto (es.: sarcoidosi avanzata, LAM). Diffusione polmonare del CO (DLCO) : ridotto Emogasanalisi : ridotta PaO2, PaCO2 Esami ematochimici selezionati (oltre agli ematochimici di routine): ANA, ENA, pANCA, cANCA (Scl-70, Jo-1, anti-DNA doppia elica, ecc) ACE Precipitine Scintigrafia polmonare con Ga-67 : può essere positivo in molte forme di pneumopatie diffuse, ma con scarsa specificità e sensibilità. Il suo uso come test in grado di monitorare l’attività della malattia è stato abbandonato per il carico radioattivo che comporta.
. Diffusione polmonare del CO (DLCO) : ridotto. Emogasanalisi : ridotta PaO2, PaCO2. Esami ematochimici selezionati (oltre agli ematochimici di routine): ANA, ENA, pANCA, cANCA (Scl-70, Jo-1, anti-DNA doppia elica, ecc) ACE. Precipitine. Scintigrafia polmonare con Ga-67 : può essere positivo in molte forme di pneumopatie diffuse, ma con scarsa specificità e sensibilità. Il suo uso come test in grado di monitorare l’attività della malattia è stato abbandonato per il carico radioattivo che comporta.")
24
HRTC : tomografia computerizzata ad alta risoluzione
Valutando analiticamente: - la distribuzione anatomica delle lesioni (regioni apicali, medie, basali, periferiche o centrali). - l’aspetto morfologico delle lesioni. è possibile una corretta diagnosi di prima-scelta nel 75-90% dei pazienti con forme importanti di pneumopatie diffuse (sarcoidosi, silicosi, IPF, ecc). I maggiori vantaggi che l’HRCT fornisce sono: riscontro precoce di lesioni polmonari non evidenziabili all’Rx del torace, possibile differenziazione di una pneumopatia diffusa da un’altra, possibile giudizio di reversibilità del processo (generalmente il riscontro di “vetro smerigliato” è a favore di reversibilità, mentre un aspetto reticolare, solitamente espressione di fibrosi, è più indicativo di irreversibilità delle lesioni, così come la presenza di cisti.
. - l’aspetto morfologico delle lesioni. è possibile una corretta diagnosi di prima-scelta nel 75-90% dei pazienti con forme importanti di pneumopatie diffuse (sarcoidosi, silicosi, IPF, ecc). I maggiori vantaggi che l’HRCT fornisce sono: riscontro precoce di lesioni polmonari non evidenziabili all’Rx del torace, possibile differenziazione di una pneumopatia diffusa da un’altra, possibile giudizio di reversibilità del processo (generalmente il riscontro di vetro smerigliato è a favore di reversibilità, mentre un aspetto reticolare, solitamente espressione di fibrosi, è più indicativo di irreversibilità delle lesioni, così come la presenza di cisti.")
25
CONNETTIVITI

26
Alveolar microlithiasis
LAM PAP

27
IPF

28
LAVAGGIO BRONCO-ALVEOLARE (BAL)
Metodica relativamente semplice e sicura che incrementa le possibilità diagnostiche delle pneumopatie diffuse senza dover ricorrere alla biopsia polmonare chirurgica; deve essere affiancato alla HRTC. Consiste nell’instillazione in un segmento broncopolmonare, durante l’indagine broncofibroscopica, di idonea quantità di soluzione fisiologica al fine di “lavare” la componenete alveolare di tale segmento; il successivo parziale recupero della soluzione mediante aspirazione permetterà di analizzare le componenti chimico-proteiche e cellulari che sono state asportate dal “polmone profondo”. Alveolite neutrofilica : IPF, asbestosi, ecc Alveolite linfocitica (CD4+/CD8+) : sarcoidosi, AAE, ecc Alveolite eosinofilica : sindr. ipereosinofila, Churg-Strauss, ecc
 : sarcoidosi, AAE, ecc. Alveolite eosinofilica : sindr. ipereosinofila, Churg-Strauss, ecc.")
29
BIOPSIA POLMONARE - Permette di fare diagnosi certa della malattia interstiziale in causa. - Pertanto, permette di impostare, laddove possibile, il trattamento corretto. Nei casi dubbi non dovrebbero essere istituiti trattamenti in grado di modificare il quadro istopatologico prima che la biopsia polmonare venga effettuata. Possibili biopsie polmonari TBLB (Trans Bronchial Lung Biopsy): biopsie piccole (sarcoidosi, altre granulomatosi, PAP, linfangiti carcinomatose). Biopsia chirurgica: toracotomica (minima), VATS. (IPF, ecc).
: biopsie piccole (sarcoidosi, altre granulomatosi, PAP, linfangiti carcinomatose). Biopsia chirurgica: toracotomica (minima), VATS. (IPF, ecc).")
31
POLMONITI INTERSTIZIALI IDIOPATICHE
Sono un gruppo di malattie interstiziali del polmone che comprende varie entità clinico-patologiche con diversi aspetti l’una dall’altra, ad etiologia sconosciuta. Classificazione A) FIBROSI POLMONARE IDIOPATICA (IPF) B) FIBROSI POLMONARI DIVERSE DALLA IPF Polmonite Interstiziale Acuta (AIP) Polmonite Interstiziale Desquamativa (DIP) Bronchiolite Respiratoria-assciata a Malattia Interstiziale Polmonare (RB-ILD) Polmonite Interstiziale Linfocitica (LIP) Polmonite Organizzativa Criptogenetica (COP) Polmonite Interstiziale Non-Specifica (NSIP)
 FIBROSI POLMONARE IDIOPATICA (IPF) B) FIBROSI POLMONARI DIVERSE DALLA IPF. Polmonite Interstiziale Acuta (AIP) Polmonite Interstiziale Desquamativa (DIP) Bronchiolite Respiratoria-assciata a Malattia Interstiziale Polmonare (RB-ILD) Polmonite Interstiziale Linfocitica (LIP) Polmonite Organizzativa Criptogenetica (COP) Polmonite Interstiziale Non-Specifica (NSIP)")
32
FIBROSI POLMONARE IDIOPATICA (IPF) (Hamman & Rich ?)
Definizione Forma specifica di pneumopatia interstiziale cronica fibrosante ad etiologia sconosciuta che richiede, dal punto di vista diagnostico, una biopsia polmonare caratterizzata da un preciso quadro istopatologico (UIP), nonché: l’esclusione di possibili altre etiologie conosciute (farmaci, esposizioni ambientali, connettiviti, ecc) prove di funzionalità respiratoria alterate (deficit restrittivo) con ipossiemia e deficit DLCO caratteristico aspetto Rx o HRCT del torace. Criteri minori età > 50 aa insorgenza subdola con dispnea durata della malattia > 3-6 mesi crepitii inspiratori (“crackles”) bibasali all’auscultazione del torace
, nonché: l’esclusione di possibili altre etiologie conosciute (farmaci, esposizioni ambientali, connettiviti, ecc) prove di funzionalità respiratoria alterate (deficit restrittivo) con ipossiemia e deficit DLCO. caratteristico aspetto Rx o HRCT del torace. Criteri minori. età > 50 aa. insorgenza subdola con dispnea. durata della malattia > 3-6 mesi. crepitii inspiratori ( crackles ) bibasali all’auscultazione del torace.")
33
Patogenesi - Processo molto complesso che coinvolgerebbe:
FIBROSI POLMONARE IDIOPATICA Epidemiologia - Più frequente nei maschi, età tra 40 e 70 aa (media 66), non presenta variazioni geografiche; prevalenza in aumento (da 3-6 a casi per ). Etiologia - Sconosciuta; è probabile che fattori multipli di rischio possano agire su predisposizione genetica (?). - Fattori di rischio (?): fumo, infezioni virali, esposizioni lavorative, antidepressivi (imipramina), reflusso gastro-esofageo. Patogenesi - Processo molto complesso che coinvolgerebbe: una primitiva fase di danno polmonare una secondaria risposta immunologica con precoce risposta citochinica e richiamo di cellule infiammatorie un eccessivo processo riparativo fibrogenetico
, non presenta variazioni geografiche; prevalenza in aumento (da 3-6 a casi per ). Etiologia - Sconosciuta; è probabile che fattori multipli di rischio possano agire su predisposizione genetica ( ). - Fattori di rischio ( ): fumo, infezioni virali, esposizioni lavorative, antidepressivi (imipramina), reflusso gastro-esofageo. Patogenesi - Processo molto complesso che coinvolgerebbe: una primitiva fase di danno polmonare. una secondaria risposta immunologica con precoce risposta citochinica e richiamo di cellule infiammatorie. un eccessivo processo riparativo fibrogenetico.")
34
Quadro eterogeneo nel tempo e nello spazio!
FIBROSI POLMONARE IDIOPATICA UIP Usual Interstitial Pneumonia Istopatologia - A piccolo ingrandimento aspetto eterogeneo caratterizzato da alternanza di zone di polmone normale, infiammazione interstiziale, fibrosi e zone a “nido d’ape”; le alterazioni sono più evidenti nelle zone più periferiche, subpleuriche del polmone. L’infiammazione interstiziale è caratterizzata da infiltrazione dei setti alveolari di linfociti e plasmacellule, con iperplasia dei pneumociti di II ordine. Le zone fibrotiche sono composte da collagene denso con presenza di “foci fibroblastici”. Quadro eterogeneo nel tempo e nello spazio!

35
FIBROSI POLMONARE IDIOPATICA
Sintomi - Dispnea da sforzo ingravescente per mesi, con possibile tosse stizzosa. Esame obiettivo - Possibile cianosi, specie dopo sforzo; frequente “ippocratismo digitale semplice”. All’auscultazione del torace, nelle fasi iniziali, presenza di rantoli crepitanti fine-inspirazione alle basi; nelle fasi avanzate possibile estensione a tutte le zone del torace e per tutta l’inspirazione. Possibile comparsa, nelle fasi avanzate, di ipertensione polmonare e cuore polmonare. Esami diagnostici - Esami ematochimici: di scarsa utilità in quanto non specifici per l’IPF; possono essere d’aiuto permettendo di escludere altre patologie (es: Ac anti- nucleari in connettiviti). - PFR: danno ventilatorio di tipo restrittivo con ridotta DLCO. - Lavaggio Broncoalveolare (BAL): reperto aspecifico mostrando un aumento della cellularità totale con neutrofilia (70-90%) e/o eosinifilia (40-60%); serve anzitutto on quanto permette d’escludere altre patologie quali infezioni, specialmente tbc, e neoplasie.
. - PFR: danno ventilatorio di tipo restrittivo con ridotta DLCO. - Lavaggio Broncoalveolare (BAL): reperto aspecifico mostrando un aumento della cellularità totale con neutrofilia (70-90%) e/o eosinifilia (40-60%); serve anzitutto on quanto permette d’escludere altre patologie quali infezioni, specialmente tbc, e neoplasie.")
36
Radiografia del torace: usualmente reticolazione/reticolo-nodulazione bibasale (60-80%), prevalente alla periferia del polmone (60%); possibile comparsa di aspetto a “nido d’ape” nelle fasi avanzate”; perdita progressiva di “volume polmonare”.
, prevalente alla periferia del polmone (60%); possibile comparsa di aspetto a nido d’ape nelle fasi avanzate ; perdita progressiva di volume polmonare .")
37
Tomografia compiuterizzata ad alta risoluzione (HRTC)
Tomografia compiuterizzata ad alta risoluzione (HRTC). Ha completamente stravolto in questi ultimi 10 anni l’approccio diagnostico a questa patologia; la sua interpretazione è strettamente correlata all’esperienza del radiologo e va valutata insieme ai dati clinici. L’HRTC può evidenziare la malattia molto prima che questa si manifesti all’Rx. L’aspetto tipico evidenzia reticolazione grossolana nelle regioni basali, posteriore nelle prime fasi, periferica/sub-pleurica. Alterazioni cistiche “a nido d’ape”e bronchiectasie divengono evidenti per la trazione fibrotica.
. Ha completamente stravolto in questi ultimi 10 anni l’approccio diagnostico a questa patologia; la sua interpretazione è strettamente correlata all’esperienza del radiologo e va valutata insieme ai dati clinici. L’HRTC può evidenziare la malattia molto prima che questa si manifesti all’Rx. L’aspetto tipico evidenzia reticolazione grossolana nelle regioni basali, posteriore nelle prime fasi, periferica/sub-pleurica. Alterazioni cistiche a nido d’ape e bronchiectasie divengono evidenti per la trazione fibrotica.")
38
FIBROSI POLMONARE IDIOPATICA
Prognosi. Infausta: 50% di mortalità a 3 anni dalla diagnosi, non solo per insufficienza respiratoria ma anche per la possibile comparsa di cardiopatia (20%), infezioni, carcinoma polmonare (10%). Non esistono indicatori sufficientemente certi che possano predire la progressione della malattia (giovane età di comparsa?, cellularità>fibrosi nelle biopsie?, effetto protettivo del fumo?). Terapia. Non vi è alcuna terapia standardizzata efficace nel trattamento dell’IPF. L’IPF è attualmente la principale condizione in cui è indicato il trapianto polmonare: trapianto di polmone singolo con sopravvivenza a 1 e 5 anni è rispettivamente 71% e 49% (* dati USA 1999).
, infezioni, carcinoma polmonare (10%). Non esistono indicatori sufficientemente certi che possano predire la progressione della malattia (giovane età di comparsa , cellularità>fibrosi nelle biopsie , effetto protettivo del fumo ). Terapia. Non vi è alcuna terapia standardizzata efficace nel trattamento dell’IPF. L’IPF è attualmente la principale condizione in cui è indicato il trapianto polmonare: trapianto di polmone singolo con sopravvivenza a 1 e 5 anni è rispettivamente 71% e 49% (* dati USA 1999).")
40
Polmoniti da ipersensibilità (AAE, Alveoliti Allergiche Estrinseche)
Definizione. Infiammazione immuno-mediata dei bronchioli terminali e alveoli indotta, in soggetti predisposti, dalla inalazione ripetuta di antigeni organici. E’ quasi sempre una malattia professionale Può essere acuta o cronica Si sviluppa solo in una piccola parte dei soggetti esposti La reazione immunologica è sempre la stessa, indipendentemente dall’antigene

41
ANTIGENE Actynomiceti termofili B. cereus Deiezioni aviarie Penicillum casei Cryptosoma corticale Sitophylus granarius MALATTIA Polmone del contadino Bissinosi, bagassosi Mal. dei condizionatori Pidgeon’s lung Mal. dei lavoratori caseari Suberosi Mal. del mugnaio

42
Polmonite da ipersensibilità
Patogenesi. L’antigene deve essere abbastanza piccolo da penetrare fino all’alveolo (generalmente < 3 di diametro), dove sviluppa appunto un’alveolite; questa è sempre la lesione iniziale. Deve esistere una predisposizione alla malattia. La reazione iniziale è di tipo III (IgG ed IgA). Raramente si evidenziano IgE specifiche per l’antigene (tranne nel caso degli isocianati MDI e TDI). Importante anche la degranulazione dei mastociti, l’attivazione dei T citotossici (CD8+) e del complemento. Se l’esposizione persiste, la flogosi diventa cronica e cellulo-mediata (IV tipo).
, dove sviluppa appunto un’alveolite; questa è. sempre la lesione iniziale. Deve esistere una predisposizione alla malattia. La reazione iniziale è di tipo III (IgG ed IgA). Raramente si evidenziano IgE specifiche per l’antigene (tranne nel caso degli isocianati MDI e TDI). Importante anche la degranulazione dei mastociti, l’attivazione dei T citotossici (CD8+) e del complemento. Se l’esposizione persiste, la flogosi diventa cronica e cellulo-mediata (IV tipo).")
43
Istopatologia Forma acuta. Alveolite linfocitaria con infiltrazione interstiziale, e alveolare, di linfociti, mastociti, macrofagi, neutrofili (polmonite interstiziale cellulare). Forma subacuta. Caratterizzata dalla presenza di granulomi senza caseosi, simili a quelli della sarcoidosi. Forma cronica. Fibrosi interstiziale
. Forma subacuta. Caratterizzata dalla. presenza di granulomi senza caseosi, simili a quelli della sarcoidosi. Forma cronica. Fibrosi interstiziale.")
44
La gravità ed il tipo di sintomi dipendono dalla durata
Polmonite da ipersensibilità La gravità ed il tipo di sintomi dipendono dalla durata dell’esposizione e dalla quantità di antigene inalato. FORMA ACUTA Esposizione massiva ed episodica Quadro simil-polmonite Quadro asmatico franco Si risolve con l’allontanamento dalla fonte antigenica FORMA CRONICA Esposizione subdola e continuata Quadro sfumato. Febbricola, malessere Evoluzione verso la fibrosi interstiziale. Insuff. respiratoria

45
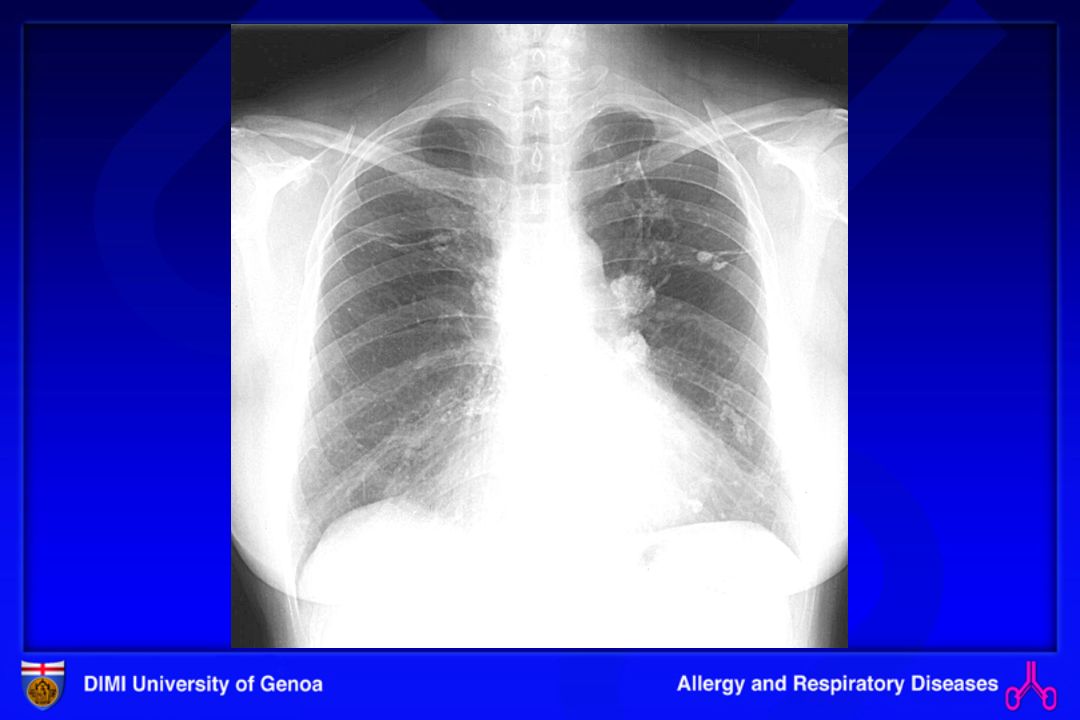
AAE: INDAGINI Rx torace.Quadro radiologico di pneumopatia interstiziale (infiltrati interstiziali e alveolari, micronodulazione nelle fasi acute; fibrosi nelle forme croniche).
.")
47
Spirometria. Quadro restrittivo e riduzione della DLCO Precipitine ???
AAE: INDAGINI BAL. Aumento della cellularità totale con linfocitosi (CD8+) e conseguente riduzione del rapporto CD4+/CD8+ nel BAL (nelle forme acute può essere presente anche neutrofilia). Spirometria. Quadro restrittivo e riduzione della DLCO Precipitine ??? Test di esposizione specifico?
 e conseguente riduzione del rapporto CD4+/CD8+ nel BAL (nelle forme acute può essere presente anche neutrofilia). Spirometria. Quadro restrittivo e riduzione della DLCO. Precipitine Test di esposizione specifico")
48
Polmoniti da ipersensibilità : criteri diagnostici
Rilievo di esposizione all’antigene documentata Anamnesi lavorativa, indagini microbiologiche Sintomi compatibili con una AAE, insorgenti dopo alcune ore dalla esposizione all’antigene aspetto radiografico compatibile fondamentali rantoli crepitanti basali alterazioni del DLCO ipossiemia e deficit ventilatorio di tipo restrittivo alveolite linfocitaria nel BAL aspetti istologici compatibili con una PI sussidiari

49
Polmonite da ipersensibilità : diagnosi differenziale
FORME ACUTE : febbre da umidificatori (inalazione di endotossine di origine batterica o fungina ) polmoniti interstiziali da virus, mycoplasmi, clamidie e rickettsie) Asma allergica FORME CRONICHE : sarcoidosi fibrosi primitive idiopatiche
 polmoniti interstiziali da virus, mycoplasmi, clamidie. e rickettsie) Asma allergica. FORME CRONICHE : sarcoidosi. fibrosi primitive idiopatiche.")
Presentazioni simili