Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Neoplasie Endocrine Multiple (MEN) Prof.ssa P. Gargiulo
 Prof.ssa P. Gargiulo")
2
MEN (Multiple Endocrine Nneoplasia) è un complesso quadro clinico caratterizzato dalla combinazione di tumori endocrini multipli e tumori neuroendocrini (NET) del duodeno, pancreas, timo, e tessuto bronchiale
 è un complesso quadro clinico caratterizzato dalla combinazione di tumori endocrini multipli e tumori neuroendocrini (NET) del duodeno, pancreas, timo, e tessuto bronchiale")
3
La malignità interviene nel 30% dei casi NETs maligni sono la causa della morbilità e mortalità correlata

4
Uno screening regolare clinico, biochimico e strumentale è alla base dell’indice prognostico. Un diagnosi precoce ed un adeguato «management» di queste neoplasie riduce il rischio di morte e la morbidità

5
MEN1 associazione di almeno 2 tumori endocrini (PTH, ipofisi, pancreas endocrino) MEN1 familiare è definita quando 1 caso di MEN1 è presente almeno in due parenti di primo grado
 MEN1 familiare è definita quando 1 caso di MEN1 è presente almeno in due parenti di primo grado")
6
Expressions of MEN1 with estimated penetrance Endocrine featuresNonendocrine features Parathyroid adenoma (90%)Lipomas (30%) Entero-pancreatic tumor (80%)Facial angiofibromas (85%) Gastrinoma (40%)Collagenomas (70%) Insulinoma (10%) NF1 including pancreatic polypeptide (20%2 ) Other: glucagonoma, VIPoma, somatostatinoma, etc. (2%) Foregut carcinoid Thymic carcinoid NF (2%)Bronchial carcinoid NF (2%) Gastric enterochromaffin-like tumor NF (10%) Anterior pituitary tumor Prolactinoma (20%) Other: GH+ PRL, GH, NF (each 5%) ACTH (2%), TSH (rare) Adrenal cortex NF (25%)
 Foregut carcinoid Thymic carcinoid NF (2%)Bronchial carcinoid NF (2%) Gastric enterochromaffin-like tumor NF (10%) Anterior pituitary tumor Prolactinoma (20%) Other: GH+ PRL, GH, NF (each 5%) ACTH (2%), TSH (rare) Adrenal cortex NF (25%).")
7
MEN2 MEN2 è una sindrome autosomica dominante in cui tutte le varianti includono un’alta penetranza del carcinoma midollare della (MTC); MEN2A è caratterizzata dalla presenza di MTC (90%), associato a feocromocitoma unilaterale o bilaterale (50%) e adenomi paratiroidei multipli (20- 30%) MEN2A rappresenta circa il 70% di MEN2 MTC è la prima manifestazione perchè a più alta penetranza;
; MEN2A è caratterizzata dalla presenza di MTC (90%), associato a feocromocitoma unilaterale o bilaterale (50%) e adenomi paratiroidei multipli (20- 30%) MEN2A rappresenta circa il 70% di MEN2 MTC è la prima manifestazione perchè a più alta penetranza;")
8
MEN2 MEN2B è più aggressiva ed è caratterizzata dalla presenza di MTC e feocromocitoma associati ad habitus marfanoide e ganglioneuromatosi mucosali ed intestinali

9
C.M. ♀ anni 34 Anamnesi patologica prossima : Inizio della sintomatologia: Inizio della sintomatologia: agosto del 1999, durante l’allattamento Sintomi e segni: Sintomi e segni: lipotimie, parestesie alla lingua e alle mani, tremori e sudorazione profusa Orario di comparsa: Orario di comparsa: metà mattina, circa 3 ore dalla colazione, prevalentemente nei periodi nei quali ha seguito diete ipocaloriche Regressione: Regressione: dopo assunzione di cibo, in genere dolci

10
INDAGINI BIOCHIMICHE

11
Glicemia 27 Glicemia 27 mg/dl (v.n.70-110) Insulinemia 48.6 Insulinemia 48.6μU/ml (v.n.2-25) C pep6.0 C pep6.0ng/ml (v.n.1-3) BG/IRI 0.5 BG/IRI 0.5(v.n.>4)
 Insulinemia 48.6 Insulinemia 48.6μU/ml (v.n.2-25) C pep6.0 C pep6.0ng/ml (v.n.1-3) BG/IRI 0.5 BG/IRI 0.5(v.n.>4)")
12
Gastrina Gastrina30 pg/ml(v.n < 40) Glucagone Glucagone144 pg/ml (v.n.60-200) PTH81.7 PTH81.7 pg/ml (v.n.10.6-54) Calcitonina Calcitonina10.1 pg/ml (v.n. < 30) Calcemia 10.21 Calcemia 10.21 mg/dl (v.n.8.5-10.2) Fosforemia Fosforemia 3.0 mg/dl(v.n.2.7-4.5) Ca ++1.54 Ca ++1.54mmol/L(v.n.1.17-1.33) Cromogranina Cromogranina75ng/ml(v.n.< 90) NSE NSE4.9ng/ml(v.n.<10)
 Calcemia Calcemia mg/dl (v.n ) Fosforemia Fosforemia 3.0 mg/dl(v.n ) Ca Ca mmol/L(v.n ) Cromogranina Cromogranina75ng/ml(v.n.< 90) NSE NSE4.9ng/ml(v.n.<10).")
13
TSH2.2 μU/ml (v.n.0.25-4) GH 0.4ng/ml (v.n.0.1-10) PRL24.5ng/ml (v.n.2.7-26) LH5.2 mU/ml (v.n.0.9-10.6) FSH6.5 mU/ml (v.n.2.5-20) FT41.0 ng/dl (v.n.0.78-1.84) FT34.6 pg/ml (v.n.1.84-5.6) IGF-1 189 ng/ml (v.n.144-360) ACTH18.8pg/ml (v.n.10-60) Cortisolo6.3 ng/dl (v.n.5.6-23.1)
 GH 0.4ng/ml (v.n ) PRL24.5ng/ml (v.n ) LH5.2 mU/ml (v.n ) FSH6.5 mU/ml (v.n ) FT41.0 ng/dl (v.n ) FT34.6 pg/ml (v.n ) IGF ng/ml (v.n ) ACTH18.8pg/ml (v.n.10-60) Cortisolo6.3 ng/dl (v.n )")
14
OGTT Tempi: (min) 0306090120 150180210240300 Glicemia (mg/dl) 42826353746629303747 Insulina (μU/ml) 11.637.14122.728.724.55.84.83.65
 Glicemia (mg/dl) Insulina (μU/ml)")
15
Prova del digiuno Tempi (ora)14°15°16°17°18°19° Glicemia (mg/dl)665551 5341 Insulina (μUI/ml)19.222.427.46.36.57.9
14°15°16°17°18°19° Glicemia (mg/dl) Insulina (μUI/ml)")
16
INDAGINI STRUMENTALI

17
TC spirale addome A carico del corpo-coda del pancreas si osservano alcune aree ipodense, contigue tra loro, a contenuto fluido-corpuscolato, di dimensioni comprese fra 0.5 e 1 cm di diametro. Nel corpo del pancreas si apprezza una raccolta “fluida” di circa 2 cm di diametro massimo; analogo reperto si apprezza anteriormente alla testa del pancreas.

18
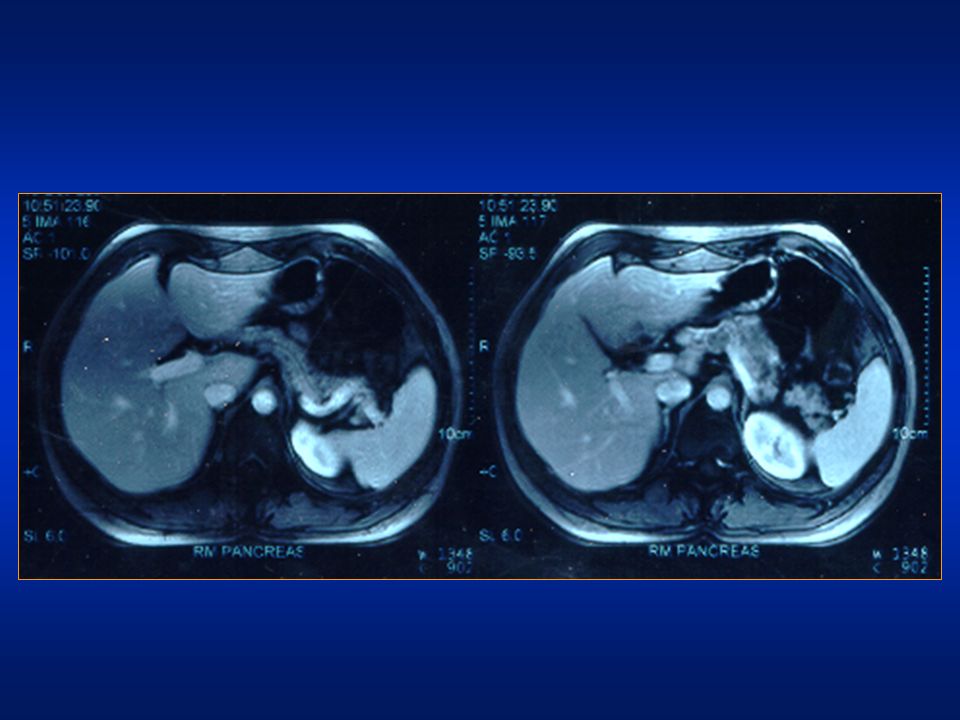
RM addome Pancreas morfovolumetricamente nei limiti. In corrispondenza dell’istmo è apprezzabile una formazione nodulare ipervascolarizzata di 18 mm. Non evidente la seconda lesione segnalata in un precedente esame TC.

20
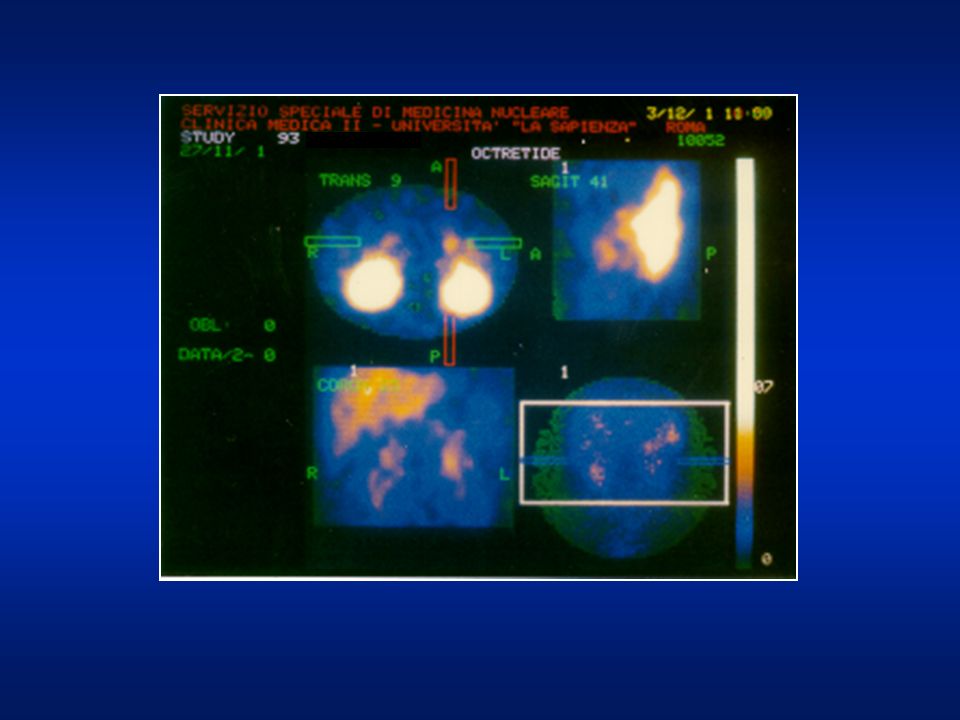
Scintigrafia con octreotide marcato L’indagine scintigrafica eseguita con esame tomografico (SPECT) dell’addome a 6 e 24 ore dall’iniezione del radiofarmaco e con elaborazione delle immagini con doppio filtro ad alta sensibilità e specificità, non ha messo in evidenza aree di patologica captazione del tracciante a livello pancreatico.
 dell’addome a 6 e 24 ore dall’iniezione del radiofarmaco e con elaborazione delle immagini con doppio filtro ad alta sensibilità e specificità, non ha messo in evidenza aree di patologica captazione del tracciante a livello pancreatico.")
22
Ecografia del collo Tiroide in sede, con dimensioni normali ed ecostruttura omogenea. Trachea in asse. Non immagini riferibili a paratiroidi aumentate di volume.

23
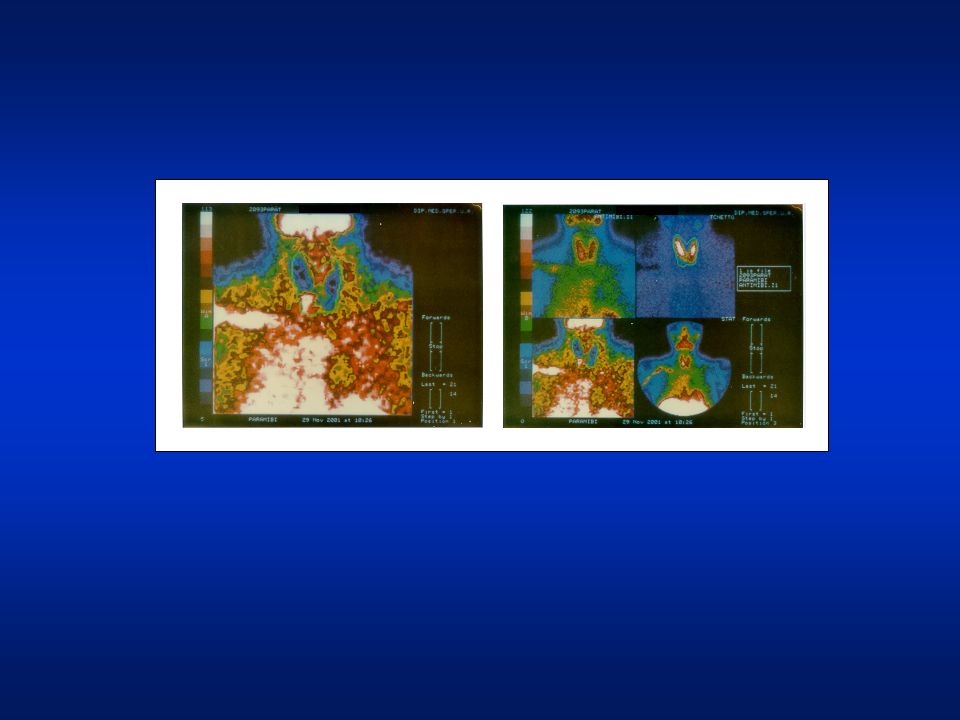
scintigrafia delle paratiroidi con doppio tracciante: 99mTc e 99mTc-MIBI Si evidenziano più aree di residua radioconcentrazione legate al sesta-MIBI: due in corrispondenza delle porzioni apicali paraistimiche di entrambi i lobi tiroidei ed una terza in sede basale destra. iperplasia” paratiroidea multipla. Conclusioni: il reperto è suggestivo di una condizione di “iperplasia” paratiroidea multipla.

25
MOC colonna BMD L1-L41.163 gr/cm3T score+ 1.06 BMD L1-L4 1.163 gr/cm3T score + 1.06

26
RM Ipofisi: negativa GASTROSCOPIA: negativa

27
Arteriografia con Calcio Gluconato L’esame mette in evidenza una “lesione” ipervascolarizzata di circa 1.5 cm a livello della regione fra testa e corpo del pancreas. Il cateterismo selettivo delle arterie: splenica, epatica propria, gastroduodenale e mesenterica superiore, dopo stimolo con calcio gluconato (0.025 mEq/Kg), dava i seguenti risultati:
, dava i seguenti risultati:.")
28
TEMPI (sec)0306090120180 Arteria Epatica Propria 38.2144.2105.152.871.260.8IRI (μU/ml) 9.610.29.97.38.47.2Cpep (ng/ml) 350303276262232298IRG (pg/ml) Arteria Gastroduodenale 25.1126.4150.910697.852.7IRI (μU/ml) 6.18.17.87.66.86.3C pep (ng/ml) 212204233208241212IRG (pg/ml) Arteria Splenica 49.455.349.356.843.944.3IRI (μU/ml) 7.48.29.36.57.99.9C pep (ng/ml) 171168112310377220IRG (pg/ml) Arteria Mesenterica 97.585.870.376.588.384.8IRI (μU/ml) 1.32013.313.111.16.7C pep (ng/ml) 157160171192217207IRG (pg/ml)
 Arteria Epatica Propria IRI (μU/ml) Cpep (ng/ml) IRG (pg/ml) Arteria Gastroduodenale IRI (μU/ml) C pep (ng/ml) IRG (pg/ml) Arteria Splenica IRI (μU/ml) C pep (ng/ml) IRG (pg/ml) Arteria Mesenterica IRI (μU/ml) C pep (ng/ml) IRG (pg/ml)")
29
Arteriografia con stimolazione selettiva del pancreas endocrino mediante calcio gluconato.

31
Caratteri istologici convenzionali Le neoplasie presentano struttura trabecolare; prive di mitosi e atipie cellulari. Caratteri immunoistochimici 1) Positività per marcatori neuroendocrini generali (cromogranina A 100% e sinaptofisina,100%). Positività per insulina (100%) e proinsulina (100%) e focale positività per α-hCG. Negatività per glucagone, PP, somatostatina. 2,3)Positività per marcatori neuroendocrini generali (cromogranina A 100%; sinaptofisina 100%). Positività per glucagone (100%); e negatività per insulina, proinsulina e PP. Insulinoma puro altamente differenziato glucagonomi ben differenziati Conclusioni: Insulinoma puro altamente differenziato associato a due glucagonomi ben differenziati
 Positività per marcatori neuroendocrini generali (cromogranina A 100% e sinaptofisina,100%). Positività per insulina (100%) e proinsulina (100%) e focale positività per α-hCG. Negatività per glucagone, PP, somatostatina. 2,3)Positività per marcatori neuroendocrini generali (cromogranina A 100%; sinaptofisina 100%). Positività per glucagone (100%); e negatività per insulina, proinsulina e PP. Insulinoma puro altamente differenziato glucagonomi ben differenziati Conclusioni: Insulinoma puro altamente differenziato associato a due glucagonomi ben differenziati.")
32
Anamnesi familiare: Nonna paternazio paterno Nonna paterna affetta da diabete mellito tipo 2; zio paterno affetto da MEN I. Padre Padre di 74 anni affetto da ipertensione arteriosa e gastrite. Madre Madre di 55 anni affetta da anemia non meglio precisata. Tre fratelli5 fratellastri sorellastra Tre fratelli (2 maschi e 1 femmina) in a.b.s; 5 fratellastri (2 maschi e 3 femmine) dei quali 4 in abs e una sorellastra sottoposta a pancreasectomia totale a 23 anni per neoplasia insulare.
 in a.b.s; 5 fratellastri (2 maschi e 3 femmine) dei quali 4 in abs e una sorellastra sottoposta a pancreasectomia totale a 23 anni per neoplasia insulare..")
33
Zio affetto da MEN 1 con adenoma delle paratiroidi, insulinomi multipli del pancreas, glucagonoma. Sorella operata a Nizza, all’età di 28 anni, di pancreasectomia totale per adenomatosi multipla del pancreas.

34
ASPETTI GENETICI Il gene della MEN I è localizzato sul braccio lungo del cromosoma 11 (11q13). La MEN I è una sindrome ereditaria autosomica dominante ad alta penetranza ad espressività variabile

35
Il prodotto del gene della MEN I è una proteina di 610 aa del peso molecolare di 67 kDa MENINACOOH NH 2 la Menina ha una capacità soppressiva sulla proliferazione cellulare

36
Agisce legandosi alla proteina Jun D MECCANISMO D’AZIONE DELLA MENINA La Jun D appartiene alla famiglia delle “Activating Proteins” ed ha un ruolo di attivatore trascrizionale JunDCOOH NH 2 JunD La Menina inibisce l’attività della Jun D e conseguentemente ha un’attività antimitotica

37
Interazione Menina-JunD Gene MEN 1 COOH NH 2 menina JunD Trascrizione

38
Interazione Menina-JunD Gene MEN 1 mutatoCOOH NH 2 menina JunD Trascrizione Proliferazione cellulare

39
a) non sense: è la più frequente (70%); comporta la comparsa di un codone di stop, induce la sintesi di una proteina tronca b) missense c) mutazioni dell’mRNA (rare) MUTAZIONI MENINA Ad oggi sono state identificate più di trecento mutazioni germinali e somatiche del gene della MEN I
 non sense: è la più frequente (70%); comporta la comparsa di un codone di stop, induce la sintesi di una proteina tronca b) missense c) mutazioni dell’mRNA (rare) MUTAZIONI MENINA Ad oggi sono state identificate più di trecento mutazioni germinali e somatiche del gene della MEN I")
40
Perdita del gene oncosoppressore sul cr.11q12-13, vicino al locus PYGM Autosomico dominante MEN 1 Sindrome di Wermer Presentazione clinica Iperplasia paratiroidi >95% Iperpara - tiroidismo Tumori endocrini duodeno/ pancreas 80-100% Adenomi ipofisari 54-65% Prlomi 14- 76% acromegalia 0-33% cushing 0-19% non funzionanti 24-27% Adenomi surrenalici Spesso asintomatici 27-36% Carcinoidi polmonari e timici 7% Carcinoidi gastrici 13-30% PPoma o non funz 80 -100% Gastrinoma 54% Insulinoma 21% Glucagonoma 3% VIPoma 1% GRFoma somatostati- noma Sindrome da carcinoide

41
INSULINOMI

42
Sintomi dell’ipoglicemia Rilascio adrenergico Sudorazione Sudorazione Agitazione Agitazione Ansia Ansia Palpitazioni Palpitazioni Astenia Astenia Tremore Tremore Fame Fame Pallore Pallore

43
Sintomi dell’ipoglicemia Neuroglicopenia Psichiatrici:Neurologici: IrritabilitàDiplopia AgitazioneCefalea Confusione mentaleAfasia NegativismoParestesie Amnesia retrogradaIncoordinazione motoria DelirioConvulsioni Parola incoerenteComa

44
Meccanismi di controregolazione Insulina IPOGLICEMIA 0’ 30’ 60’ Catecolamine Glucagone GH Cortisolo Glicemia

45
EPIDEMIOLOGIA INSULINOMI L'insulinoma è la neoplasia endocrina del pancreas più frequente nel 5% dei casi è associato a MEN-1 Età di insorgenza: prevalentemente tra la terza e la sesta decade Sesso: uguale frequenza negli uomini e nelle donne Localizzazione pancreas: coda 40%, corpo 28%, testa 32%. Nel 90% dei casi si tratta di neoplasie benigne, singole, di dimensioni comprese tra 1 e 2 cm di diametro Nel 90% dei casi si tratta di neoplasie benigne, singole, di dimensioni comprese tra 1 e 2 cm di diametro.

46
Anatomia patologica In base al tipo di granuli presenti nelle cellule tumorali, distinguiamo quattro tipi istologici di insulinomi cellule tipiche con granuli caratteristici cristalliformi contenenti insulinacellule tipiche con granuli caratteristici cristalliformi contenenti insulina cellule con granuli tipici che atipicicellule con granuli tipici che atipici cellule con granuli atipicicellule con granuli atipici cellule prive di granuli secretori.cellule prive di granuli secretori.

47
GRUPPO A cellule tipiche, ricche di granulazioni, organizzate in forma trabecolare, con l'insulina uniformemente distribuita all'interno del citoplasma e che rispondono, in maniera adeguata, alle prove di stimolazione o di soppressione della secrezioneinsulinica cellule tipiche, ricche di granulazioni, organizzate in forma trabecolare, con l'insulina uniformemente distribuita all'interno del citoplasma e che rispondono, in maniera adeguata, alle prove di stimolazione o di soppressione della secrezione insulinica GRUPPO B poche cellule ben granulate, struttura istologica di tipo midollare, irregolare distribuzione dei granuli di insulina e ridotta capacità di risposta ai farmaci che inibiscono la secrezione insulinica

48
Con metodi immunoistochimici è possibile dimostrare che queste cellule sono in grado di esprimere MARCATORI DI DIFFERENZIAZIONE NEUROENDOCRINA : enolasi neurono-specifica (NSE), la PGP 9.5, la sinaptofisina e la cromogranina A. MARCATORI CELLULARI cromogranina, HSL-19, insulina e proinsulina

49
DIAGNOSI

50
DIAGNOSI DI LABORATORIO si basa sulla dimostrazione di livelli plasmatici di insulina inappropriatamente elevati rispetto ai bassi valori glicemici Sintomi ipoglicemici presenti principalmente a digiuno Glicemia < 45 mg/dl Insulinemia >6 U/ml Peptide C >1 ng/ml BG/IRI < 2 indicano una condizione di inappropriata secrezione insulinica.

51
DIAGNOSI DIFFERENZIALE IPOGLICEMIA REATTIVA: i sintomi si presentano a distanza di 2-5 ore dall'assunzione di un pasto, IPOGLICEMIA FATTIZIA: assunzione accidentale o più spesso volontaria di insulina o antidiabetici orali. insulinoma insulina sulfoniluree Anticorpi anti IRI Anticorpi anti IRI assenti presenti assenti Sulfoniluree Sulfonilureeassenti assenti presenti Insulinemia Insulinemia C-peptide C-peptide ridotto Proinsulina Proinsulina normale _____________________________________________________ ______

52
PROVE DIAGNOSTICHE SoppressioneStimolo DiazossidoGlucosio SomatostatinaTolbutamide InsulinaAminoacidi GlucagoneCalcio

53
DIAGNOSTICA PER IMMAGINI

54
Ecografia specificità dal 15 al 30% difficile identificazione della ghiandola difficile identificazione della ghiandola 90% dei casi lesione ipoecogena rispetto al normale parenchima pancreatico, tondeggiante o ovalare, contorni netti e regolari 90% dei casi lesione ipoecogena rispetto al normale parenchima pancreatico, tondeggiante o ovalare, contorni netti e regolari 10% dei casi formazione iso o iper-ecogena, ecostruttura omogenea, priva di orletto perilesionale 10% dei casi formazione iso o iper-ecogena, ecostruttura omogenea, priva di orletto perilesionale

55
Ecografia intraoperatoria sensibilità : 100% se associata alla palpazione chirurgica limiti: piccole dimensioni della neoplasia limiti: piccole dimensioni della neoplasia. falsi positivi: noduli cicatriziali o da isole di tessuto pancreatico ectopico nel duodeno.

56
Tomografia Computerizzata Specificità:elevata Specificità: elevata Falsi positivi:rari Falsi positivi: rari (dovuti a noduli reattivi o a segmenti isolati di vasi tortuosi)
")
57
TC Spirale Vantaggi: Vantaggi: consente di diminuire i tempi di scansione permettendo di studiare con maggiore attenzione gli organi addominali riducendo le conseguenze dovute ai movimenti degli atti respiratori. Angio TC consiste nell'iniezione intrarteriosa di mezzo di contrasto tramite un catetere posizionato nell'arteria gastrica sinistra o mesenterica superiore Vantaggi: solo nei casi relativamente rari di tumori ipervascolarizzati Svantaggi: maggiore invasività e artefatti in prossimità del catetere.

58
Angiografia Sensibilità:63-90% dei casi soprattutto se effettuata con cateterismo selettivo o superselettivo delle arterie del distretto pancreatico Sensibilità: 63-90% dei casi soprattutto se effettuata con cateterismo selettivo o superselettivo delle arterie del distretto pancreatico falsi negativi: lesioni inferiori a 1 cm di diametro (possono essere mascherate dalle anse intestinali, dal meteorismo e dalle formazioni vascolari) falsi negativi : lesioni inferiori a 1 cm di diametro (possono essere mascherate dalle anse intestinali, dal meteorismo e dalle formazioni vascolari) Svantaggi: invasività Indicazioni: solo ai casi che presentano un forte sospetto clinico e risultati dubbi o persistentemente negativi con le metodiche non invasive Indicazioni: solo ai casi che presentano un forte sospetto clinico e risultati dubbi o persistentemente negativi con le metodiche non invasive.
 falsi negativi : lesioni inferiori a 1 cm di diametro (possono essere mascherate dalle anse intestinali, dal meteorismo e dalle formazioni vascolari) Svantaggi: invasività Indicazioni: solo ai casi che presentano un forte sospetto clinico e risultati dubbi o persistentemente negativi con le metodiche non invasive Indicazioni: solo ai casi che presentano un forte sospetto clinico e risultati dubbi o persistentemente negativi con le metodiche non invasive.")
59
Immagine arteriografica di un insulinoma della testa del pancreas

60
Arteriografia con Calcio Gluconato e cateterismo delle vene sovraepatiche (ASVS) I cateteri vengono posizionati nella vena sovraepatica attraverso la puntura della vena femorale. Dopo cateterizzazione dell'arteria femorale viene effettuata un'arteriografia pancreatica standard con iniezione selettiva di mezzo di contrasto nelle arterie gastroduodenale, splenica, epatica propria e mesenterica superiore. Seguendo ogni arteriogramma, il calcio gluconato, alla dose di 0.025 mEq Ca++/Kg diluito in 5 cc di soluzione fisiologica, viene iniettato in bolo attraverso il catetere posizionato in ognuna delle arterie selettivamente cateterizzate. Prima di ciascun bolo deve essere eseguito un prelievo basale per insulinemia e peptide C e, dopo ciascuna iniezione di calcio gluconato, vengono eseguiti i prelievi venosi dalla vena sovraepatica sinistra o destra, cateterizzate per via femorale, ad intervalli di tempo ben definiti: 30, 60, 90, 120 e 180 secondi.

61
Arteriografia con stimolazione selettiva del pancreas endocrino mediante calcio gluconato in paziente con insulinoma localizzato nella testa del pancreas; vedi valore della arteria mesenterica superiore)
")
62
Risonanza magnetica Rispetto alla TC più bassa risoluzione spaziale (fattore importante nelle lesioni di piccole dimensioni), ma una risoluzione di contrasto più elevata, da cui risulta un vantaggio nella discriminazione della zona alterata rispetto al parenchima sano
, ma una risoluzione di contrasto più elevata, da cui risulta un vantaggio nella discriminazione della zona alterata rispetto al parenchima sano")
63
Presenza di processo espansivo rotondeggiante (2 cm) a carico del versante posteriore del pancreas in corrispondenza del tratto di passaggio tra corpo e coda dell'organo.
 a carico del versante posteriore del pancreas in corrispondenza del tratto di passaggio tra corpo e coda dell organo.")
64
Scintigrafia con octreotide marcato con Indio 111 (Octreoscan) molti insulinomi mostrano la presenza di siti di legame ad alta affinità sia per SS-14 e SS-28 che per l’octreotide. La dose preferibilmente utilizzata di In111-Octreotide è di circa 200MBq. La bassa percentuale di rilevamento (46%) di questo tipo di neoplasia pancreatica è correlata alla bassa incidenza del sottotipo II dei recettori per la SS sulle cellule dell’insulinoma.
 di questo tipo di neoplasia pancreatica è correlata alla bassa incidenza del sottotipo II dei recettori per la SS sulle cellule dell’insulinoma..")
65
CONCLUSIONI DIAGNOSTICHE L’approccio ottimale alla resezione chirurgica dell’insulinoma, in termini di diagnosi preoperatoria richiede quattro stadi identificazione preoperatoria del tumore e delle regioni pancreatiche coinvolte tramite procedure d’immagine convenzionaliidentificazione preoperatoria del tumore e delle regioni pancreatiche coinvolte tramite procedure d’immagine convenzionali localizzazione del tumore tramite ASVSlocalizzazione del tumore tramite ASVS conferma della localizzazione tumorale tramite ecografia intraoperatoriaconferma della localizzazione tumorale tramite ecografia intraoperatoria palpazione durante intervento chirurgico della regione sospetta e dell’intero pancreaspalpazione durante intervento chirurgico della regione sospetta e dell’intero pancreas

66
SINDROMI DA NEOPLASIE ENDOCRINE MULTIPLE (MEN)
")
67
L'iperparatiroidismo primitivo è presente in almeno il 90% dei pazienti affetti. La manifestazione più comune è l'ipercalcemia asintomatica; circa il 25% dei pazienti mostra evidenza di nefrolitiasi o nefrocalcinosi. L'iperplasia diffusa o gli adenomi multipli si osservano più frequentemente degli adenomi solitari. Solo 1-2% degli adenomi delle paratiroidi appartengono alla MEN I. All’iperPTH spesso è associata la S. Zollinger- Ellison a causa dell’ipergastrinemia da ipercalcemia. PARATIROIDI Diagnosi: dosaggio Ca, P, Ca ionizzato, PTH scintigrafia con Sestamibi e Tecnezio (con sottrazione di immagini) Terapia: paratiroidectomia subtotale con autotrapianto.
 Terapia: paratiroidectomia subtotale con autotrapianto..")
68
TUMORI PANCREATICI I tumori delle cellule insulari pancreatiche sono stati riferiti in una percentuale variabile dal 30 al 75% dei pazienti affetti. Tumori cellule BETA: 40% origina dalle cellule ß, si manifesta prima dei 40 anni, secerne insulina ed è associato a ipoglicemia a digiuno (INSULINOMA). Triade di Whipple: Sintomi di ipoglicemia Glicemia <50 mg/dL negli uomini oppure <45 mg/dL nelle donne Regressione dei sintomi dopo l’innalzamento dei livelli plasmatici di glucosio Diagnosi: Test del digiuno La diagnosi di tumore pancreatico a cellule ß insulino-secernenti viene stabilita dimostrando la presenza di ipoglicemia a digiuno congiuntamente a elevati livelli plasmatici di insulina. Il rapporto glicemia/insulinemia inferiore a 2 indica un’inappropiata secrezione ormonale di insulina, anche il C-peptide >1 ng/ml in presenza di glicemia < 45 mg/dl è diagnostico.
. Triade di Whipple: Sintomi di ipoglicemia Glicemia <50 mg/dL negli uomini oppure <45 mg/dL nelle donne Regressione dei sintomi dopo l’innalzamento dei livelli plasmatici di glucosio Diagnosi: Test del digiuno La diagnosi di tumore pancreatico a cellule ß insulino-secernenti viene stabilita dimostrando la presenza di ipoglicemia a digiuno congiuntamente a elevati livelli plasmatici di insulina. Il rapporto glicemia/insulinemia inferiore a 2 indica un’inappropiata secrezione ormonale di insulina, anche il C-peptide >1 ng/ml in presenza di glicemia < 45 mg/dl è diagnostico..")
69
Tumori a cellule NON BETA: In circa il 60% dei casi, i tumori delle insule pancreatiche derivano da elementi cellulari non ß. I tumori a cellule non ß sono piu’ comuni nei pazienti al di sopra dei 40 anni. 1) GASTRINOMI: La gastrina è l'ormone più comunemente secreto dai tumori a cellule non ß ed è associata a ulcera peptica intrattabile e complicata (sindrome di Zollinger-Ellison). Più del 50% dei pazienti affetti da MEN I soffre di malattia ulcerosa peptica; nella maggior parte dei casi le ulcere sono multiple o a localizzazione atipica e l'incidenza di emorragia, perforazione e ostruzione è corrispondentemente alta. Sono stati identificati anche gastrinomi del duodeno, stomaco, ilo splenico. Quando i pazienti la cui presentazione clinica iniziale è la sindrome di Zollinger- Ellison vengono valutati a distanza, una percentuale variabile dal 20 al 60% si dimostra affetta dalla sindrome MEN I. DIAGNOSI: gastrinemia > 1000 pg/ml; test di stimolo con secretina, localizzazione con ecografia, TC addominale o angiografia, Octreoscan TERAPIA: Il trattamento dei tumori a cellule non ß gastrino-secernenti è complesso. In tutti i pazienti deve essere fatto un tentativo per localizzare e asportare il tumore. Se ciò è impossibile, un inibitore della pompa protonica, può frequentemente alleviare la sintomatologia dell'ulcera peptica. Gli anti-H2 possono essere utilizzati anch'essi, ma sono meno efficaci.
 GASTRINOMI: La gastrina è l ormone più comunemente secreto dai tumori a cellule non ß ed è associata a ulcera peptica intrattabile e complicata (sindrome di Zollinger-Ellison). Più del 50% dei pazienti affetti da MEN I soffre di malattia ulcerosa peptica; nella maggior parte dei casi le ulcere sono multiple o a localizzazione atipica e l incidenza di emorragia, perforazione e ostruzione è corrispondentemente alta. Sono stati identificati anche gastrinomi del duodeno, stomaco, ilo splenico. Quando i pazienti la cui presentazione clinica iniziale è la sindrome di Zollinger- Ellison vengono valutati a distanza, una percentuale variabile dal 20 al 60% si dimostra affetta dalla sindrome MEN I. DIAGNOSI: gastrinemia > 1000 pg/ml; test di stimolo con secretina, localizzazione con ecografia, TC addominale o angiografia, Octreoscan TERAPIA: Il trattamento dei tumori a cellule non ß gastrino-secernenti è complesso. In tutti i pazienti deve essere fatto un tentativo per localizzare e asportare il tumore. Se ciò è impossibile, un inibitore della pompa protonica, può frequentemente alleviare la sintomatologia dell ulcera peptica. Gli anti-H2 possono essere utilizzati anch essi, ma sono meno efficaci..")
70
2) GLUCAGONOMA: Tumore a partenza dalle cellule alfa pancreatiche, sporadico (80%), MEN I (20%), f>m, Clinica: rash cutanea da eritema nercrolitico migrante, anemia e diabete mellito. Diagnosi: Glucagonemia: 500-1000 pg/ml Localizzazione: TAC, angiografia Terapia: chirurgia (prima scelta) analoghi della somatostatina. 3) VIPOMA: Clinica: grave diarrea secretoria che porta a deplezione di liquidi e di elettroliti. Questo complesso sintomatologico è denominato sindrome WDHA (da Watery Diarrhea, Hypokalemia e Achlorydria, cioè diarrea acquosa, ipokaliemia e acloridria) o colera pancreatico.
 analoghi della somatostatina. 3) VIPOMA: Clinica: grave diarrea secretoria che porta a deplezione di liquidi e di elettroliti. Questo complesso sintomatologico è denominato sindrome WDHA (da Watery Diarrhea, Hypokalemia e Achlorydria, cioè diarrea acquosa, ipokaliemia e acloridria) o colera pancreatico..")
71
Tumori ipofisari Sono stati osservati in una percentuale variabile dal 50 al 65% dei pazienti con sindrome MEN I. Il 2% di tutti i tumori ipofisari è associato alla MEN I 2/3 sono microadenomi Circa il 25% di questi tumori secerne GH oppure GH/PRL. Le persone colpite hanno acromegalia, la quale è clinicamente indistinguibile dalla forma sporadica della malattia. Alcune segnalazioni indicano che dal 25 al 90% dei tumori secerne solo PRL. Il 3% circa secerne ACTH. La maggior parte dei rimanenti è non funzionante. La diffusione locale del tumore può causare disturbi visivi e cefalea nonché ipopituitarismo.

72
L'età all'esordio è stata descritta come variabile dai 4 agli 81 anni, ma la diagnosi è più comune tra il terzo e il quinto decennio di vita. Nei familiari di I grado, lo screening genetico mediante analisi del DNA con la tecnica del polimorfismo della lunghezza dei frammenti di restrizione è oggi in grado di identificare lo stato di portatore con un'accuratezza del 99,5%. Lo screening periodico dei portatori genetici deve essere eseguito ogni anno a partire dai 15 anni di età e deve comprendere i punti seguenti: esame retrospettivo della storia clinica del paziente per la presenza di sintomi suggestivi di malattia ulcerosa peptica, diarrea, nefrolitiasi, ipoglicemia e ipopituitarismo; esame obiettivo per evidenziare difetti del campo visivo, segni di acromegalia e lipomi sottocutanei; dosaggio del Ca sierico, dell'ormone paratiroideo integro, della gastrina e della prolattina. Se indicati, devono essere eseguiti anche esami di laboratorio e test diagnostici aggiuntivi e una TC o una RMN della sella turcica. VALUTAZIONE CLINICA E SCREENING GENETICO DELLA MEN I:

73
VARIANTE BURIN DELLA MEN I: PROLATTINOMI IPERPARATIROIDISMO RIDUZIONE DELLA PENETRANZA DEI GASTRINOMI

74
Neoplasie endocrine multiple di tipo II MEN II a: adenomatosi endocrina multipla di tipo II (sindrome di Sipple) Carcinoma Midollare della Tiroide, Iperparatiroidismo, Feocromocitoma MEN II b: adenomatosi endocrina multipla di tipo IIB; (sindrome dei neurinomi mucosi) Carcinoma Midollare della Tiroide, Feocromocitoma, Ganglioneuromatosi
 Carcinoma Midollare della Tiroide, Iperparatiroidismo, Feocromocitoma MEN II b: adenomatosi endocrina multipla di tipo IIB; (sindrome dei neurinomi mucosi) Carcinoma Midollare della Tiroide, Feocromocitoma, Ganglioneuromatosi")
75
Studi genetici recenti hanno consentito di mappare i difetti genetici presenti nella MEN IIA, nella MEN IIB, nel carcinoma midollare familiare della tiroide e nella malattia di Hirschsprung a livello della regione pericentromerica del cromosoma 10 e hanno permesso di identificare le mutazioni in un proto-oncogene che codifica per una specifica tirosin chinasi recettoriale, il ret, suggerendo che questo oncogene dominante sia responsabile delle alterazioni associate con queste condizioni. La MEN II ha una prevalenza di 1-10/100.000 Può presentarsi in forma familiare o sporadica

76
MEN II A Le caratteristiche cliniche della MEN IIA dipendono dal tipo di tumore presente. 1) CARCINOMA MIDOLLARE DELLA TIROIDE (CMT) Sporadico: 70-80% Familiare: 20% (Associato alla MEN II A 65%, MEN II B 25%, FCMT 20%) Il 65% dei pazienti affetti da carcinoma midollare della tiroide hanno una MEN II A, mentre il CMT è presente nella quasi totalità dei pazienti con MEN II A; Compare nell’età giovane-adulta, è meno aggressivo rispetto alla MEN II B Può essere multicentrico, coinvolgendo entrambi i lobi Il carcinoma midollare della tiroide viene diagnosticato misurando la calcitonina plasmatica dopo infusione provocativa di pentagastrina e Ca. Nella maggior parte dei pazienti con lesioni tiroidee palpabili i livelli basali della calcitonina sono elevati; nelle fasi precoci della malattia i livelli basali possono essere normali e il carcinoma midollare può essere diagnosticato soltanto grazie a un'esagerata risposta al Ca e alla pentagastrina.
 CARCINOMA MIDOLLARE DELLA TIROIDE (CMT) Sporadico: 70-80% Familiare: 20% (Associato alla MEN II A 65%, MEN II B 25%, FCMT 20%) Il 65% dei pazienti affetti da carcinoma midollare della tiroide hanno una MEN II A, mentre il CMT è presente nella quasi totalità dei pazienti con MEN II A; Compare nell’età giovane-adulta, è meno aggressivo rispetto alla MEN II B Può essere multicentrico, coinvolgendo entrambi i lobi Il carcinoma midollare della tiroide viene diagnosticato misurando la calcitonina plasmatica dopo infusione provocativa di pentagastrina e Ca. Nella maggior parte dei pazienti con lesioni tiroidee palpabili i livelli basali della calcitonina sono elevati; nelle fasi precoci della malattia i livelli basali possono essere normali e il carcinoma midollare può essere diagnosticato soltanto grazie a un esagerata risposta al Ca e alla pentagastrina..")
77
2) FEOCROMOCITOMA: Tumore derivante dal tessuto cromaffine del sistema nervoso simpatico che produce NORADRENALINA e/o ADRENALINA Le crisi ipertensive rappresentano il quadro classico della malattia e si associano a sintomi cardiovascolari e vasomotori (cefalea, sudorazione, cardiopalmo,tachicardia, pallore, nausea e vomito, ansia) Nella forma associata alla MEN II si manifesta come multifocale e bilaterale nel 65-90% dei pazienti. Nel 25 % dei pazienti è maligno, e raramente si localizza in sede extra- surrenalica 10% in età giovanile Fino al 50% negli anziani

78
Poiché il feocromocitoma può essere asintomatico nei pazienti con MEN IIA, può essere difficile escluderlo con certezza. Il metodo più sensibile per porre diagnosi di MEN IIA è il dosaggio delle catecolamine libere in un campione di urine delle 24 h e acido vanilmandelico (VMA). L'escrezione di acido vanilmandelico è spesso normale nelle prime fasi della malattia. La TC o la RMN sono utili ai fini della localizzazione del feocromocitoma o dell'accertamento della presenza di lesioni bilaterali. È improbabile che la scintigrafia esterna con 131I-metaiodobenzilguanidina fornisca informazioni aggiuntive.
. L escrezione di acido vanilmandelico è spesso normale nelle prime fasi della malattia. La TC o la RMN sono utili ai fini della localizzazione del feocromocitoma o dell accertamento della presenza di lesioni bilaterali. È improbabile che la scintigrafia esterna con 131I-metaiodobenzilguanidina fornisca informazioni aggiuntive..")
79
3) IPERPARATIROIDISMO Meno comune del carcinoma midollare della tiroide o del feocromocitoma. Circa il 25% dei pazienti colpiti in una linea familiare con MEN IIA presenta evidenza clinica di iperparatiroidismo (che può essere presente da molto tempo), con ipercalcemia, nefrolitiasi, nefrocalcinosi o insufficienza renale. In un ulteriore 25%, senza evidenza clinica o biochimica di iperparatiroidismo, l'iperplasia delle paratiroidi viene scoperta incidentalmente durante il trattamento chirurgico del carcinoma midollare della tiroide. Come nella sindrome MEN I, l'iperparatiroidismo spesso coinvolge più di una ghiandola, come iperplasia diffusa o come adenomi multipli.
, con ipercalcemia, nefrolitiasi, nefrocalcinosi o insufficienza renale. In un ulteriore 25%, senza evidenza clinica o biochimica di iperparatiroidismo, l iperplasia delle paratiroidi viene scoperta incidentalmente durante il trattamento chirurgico del carcinoma midollare della tiroide. Come nella sindrome MEN I, l iperparatiroidismo spesso coinvolge più di una ghiandola, come iperplasia diffusa o come adenomi multipli..")
80
VALUTAZIONE CLINICA E SCREENING GENETICO Lo screening genetico per la MEN IIA può adesso essere eseguito con un alto grado di accuratezza. Nei portatori genetici identificati, in base al codone mutato del gene RET è consigliabile eseguire una tiroidectomia profilattica durante la prima infanzia o all'inizio della seconda infanzia (stratificazione del rischio genetico o COPS: codon oriented prophylactic surgery) perché il carcinoma midollare della tiroide risulta fatale se non trattato. Uno screening annuale per l'iperparatiroidismo e il feocromocitoma deve cominciare presto durante l'infanzia e proseguire indefinitamente. La storia clinica del paziente deve essere riesaminata alla ricerca di sintomi indicativi di feocromocitoma (cefalea parossistica, sudorazione o palpitazioni) e di coliche renali. Deve essere controllata la PA e devono essere eseguite indagini di laboratorio. Nei pazienti che non sono stati sottoposti a tiroidectomia profilattica, la diagnosi precoce di carcinoma midollare della tiroide rimane di importanza fondamentale, in modo che il tumore possa essere rimosso quando è ancora localizzato all'interno della ghiandola.
 perché il carcinoma midollare della tiroide risulta fatale se non trattato. Uno screening annuale per l iperparatiroidismo e il feocromocitoma deve cominciare presto durante l infanzia e proseguire indefinitamente. La storia clinica del paziente deve essere riesaminata alla ricerca di sintomi indicativi di feocromocitoma (cefalea parossistica, sudorazione o palpitazioni) e di coliche renali. Deve essere controllata la PA e devono essere eseguite indagini di laboratorio. Nei pazienti che non sono stati sottoposti a tiroidectomia profilattica, la diagnosi precoce di carcinoma midollare della tiroide rimane di importanza fondamentale, in modo che il tumore possa essere rimosso quando è ancora localizzato all interno della ghiandola..")
81
Terapia In un paziente che si presenta sia con feocromocitoma sia con carcinoma midollare della tiroide o iperparatiroidismo, il feocromocitoma deve essere rimosso per primo perché, anche se asintomatico, esso aumenta notevolmente il rischio dell'intervento chirurgico per il carcinoma midollare della tiroide o per l'iperparatiroidismo. La chemioterapia si è dimostrata inefficace per il trattamento del carcinoma midollare della tiroide metastatico o residuo, mentre la radioterapia può prolungare la sopravvivenza.

82
Neoplasie endocrine multiple di tipo IIb (men IIb) (MEN III o adenomatosi endocrina multipla di tipo IIB o sindrome dei neurinomi mucosi) Sindrome caratterizzata dalla presenza di neurinomi mucosi multipli, carcinoma midollare della tiroide e feocromocitoma, spesso associati con habitus marfanoide. La reale incidenza della sindrome MEN IIB sporadica è sconosciuta. A differenza che nella MEN I e nella MEN IIA, l'iperparatiroidismo si osserva raramente nella MEN IIB. Nei pazienti con MEN IIB studi genetici hanno identificato mutazioni a carico dell'oncogene ret localizzato sul cromosoma 10.

83
Sintomi e segni La caratteristica distintiva della sindrome MEN IIB è la presenza di neurinomi mucosi nella maggior parte dei soggetti colpiti. I neurinomi appaiono come piccoli rigonfiamenti lucenti a livello delle labbra, della lingua e della mucosa orale. Anche le palpebre, la congiuntiva e la cornea vengono frequentemente interessate. Sono caratteristici l'ispessimento delle palpebre e la diffusa ipertrofia delle labbra. I disturbi GI legati all'alterazione della motilità (stipsi, diarrea e, occasionalmente, megacolon) sono di comune riscontro e si pensa che siano causati da una ganglioneuromatosi intestinale diffusa. Sebbene i neurinomi, le caratteristiche facciali e i disturbi GI siano presenti già in giovane età, la sindrome spesso non viene riconosciuta fino al manifestarsi di un carcinoma midollare della tiroide o di un feocromocitoma, in età più avanzata. Oltre all'habitus marfanoide, sono comuni le anomalie scheletriche della colonna vertebrale (lordosi, cifosi, scoliosi), il piede cavo e il piede talo-equino-varo. Circa la metà dei casi descritti presenta la sindrome completa con neurinomi mucosi, feocromocitomi e carcinoma midollare della tiroide. Meno del 10% dei pazienti presenta solo neurinomi e feocromocitomi, mentre i rimanenti hanno neurinomi e carcinoma midollare della tiroide senza feocromocitoma. (Il carcinoma midollare della tiroide tende a essere particolarmente aggressivo nella MEN IIB e può essere presente nei bambini molto piccoli).
 sono di comune riscontro e si pensa che siano causati da una ganglioneuromatosi intestinale diffusa. Sebbene i neurinomi, le caratteristiche facciali e i disturbi GI siano presenti già in giovane età, la sindrome spesso non viene riconosciuta fino al manifestarsi di un carcinoma midollare della tiroide o di un feocromocitoma, in età più avanzata. Oltre all habitus marfanoide, sono comuni le anomalie scheletriche della colonna vertebrale (lordosi, cifosi, scoliosi), il piede cavo e il piede talo-equino-varo. Circa la metà dei casi descritti presenta la sindrome completa con neurinomi mucosi, feocromocitomi e carcinoma midollare della tiroide. Meno del 10% dei pazienti presenta solo neurinomi e feocromocitomi, mentre i rimanenti hanno neurinomi e carcinoma midollare della tiroide senza feocromocitoma. (Il carcinoma midollare della tiroide tende a essere particolarmente aggressivo nella MEN IIB e può essere presente nei bambini molto piccoli)..")
84
Diagnosi e terapia Le implicazioni per la diagnosi, lo screening familiare e la terapia sono le stesse per la sindrome MEN IIA. Lo screening genetico per la MEN IIB può adesso essere eseguito con un elevato grado di accuratezza. Nei portatori genetici identificati è consigliabile eseguire una tiroidectomia profilattica nel periodo neonatale o nella prima infanzia e tutti i pazienti affetti devono essere sottoposti a tiroidectomia totale non appena sia stata stabilita la diagnosi. Il feocromocitoma, se presente, deve essere asportato prima di eseguire la tiroidectomia.

Presentazioni simili



>")











>")



 e sull’ equilibrio.>")
