Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

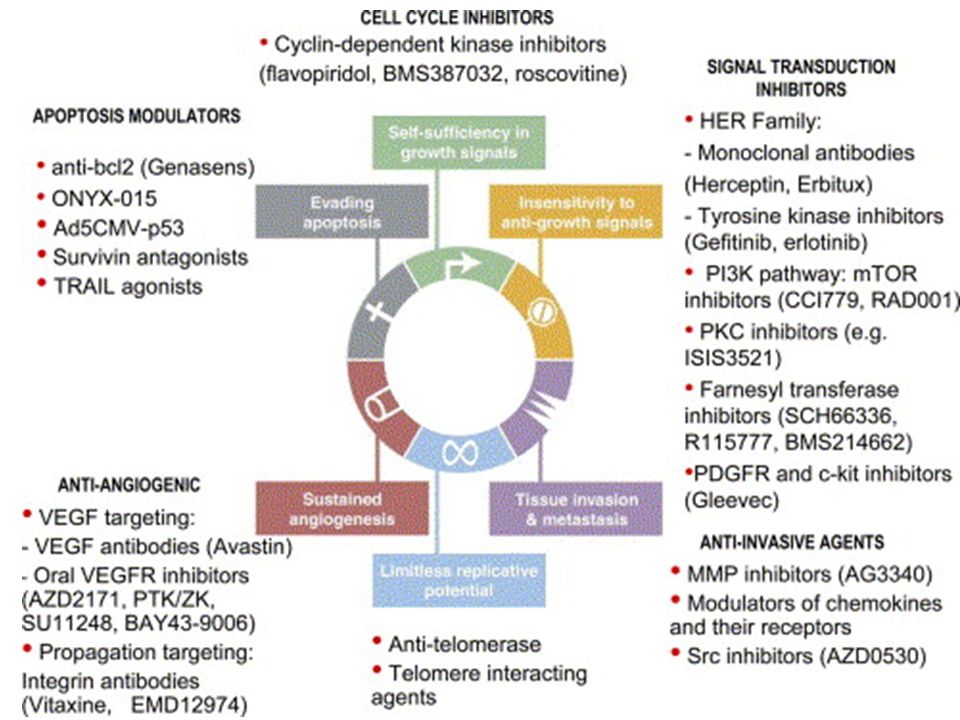
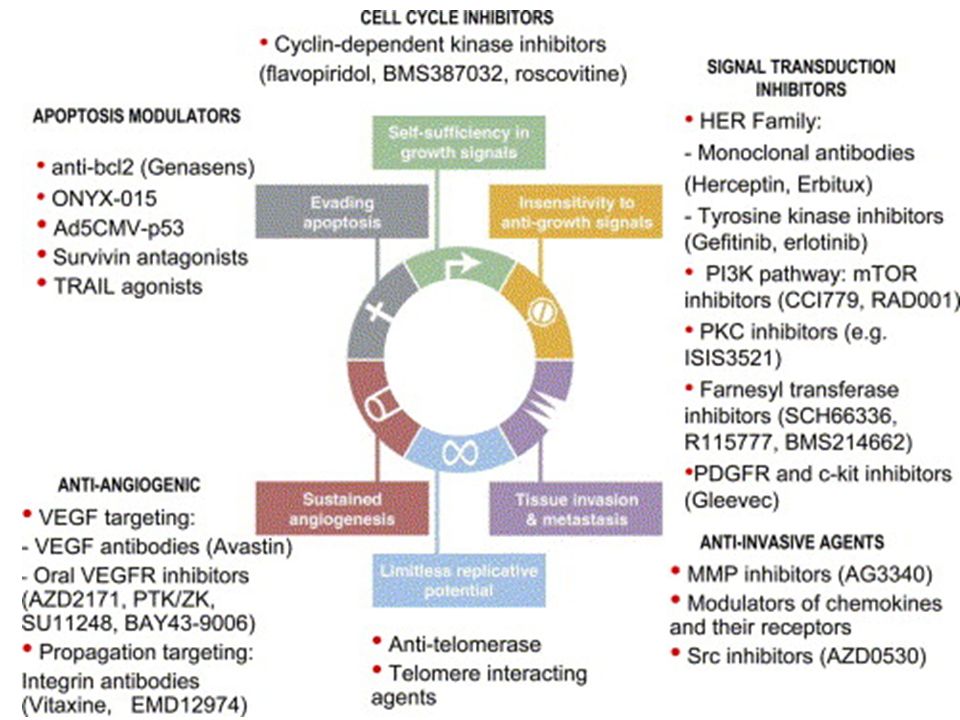
2
Perché usare farmaci bersaglio-specifici?

3
FINESTRA TERAPEUTICA RISTRETTA SVILUPPO DI RESISTENZA EFFICACIA LIMITATA

5
L’ANGIOGENESI È UN PROCESSO ESSENZIALE ALLO SVILUPPO TUMORALE
Fattori di crescita Tumore While there are more than 100 distinct types of cancer (and considerable heterogeneity within each tumor type), there exists a remarkable similarity in the pathologic traits that collectively drive tumor growth. Across most—if not all—malignancies, sustained angiogenesis is considered to be one of these central cancer “hallmarks.”1 The image shown here depicts pathologic angiogenesis. To grow beyond 1 to 2 mm in diameter, a tumor needs an independent blood supply, which is acquired by expressing growth factors that recruit new vasculature from existing blood vessels. This process continues even as the tumor matures. Angiogenesis has been correlated with disease progression and/or poor prognosis in many tumor types—including lung, colon, breast, renal, and other cancers—and can be activated at different stages of tumor development, depending on the tumor type and microenvironmental conditions.2-5 Vasi sanguigni Angiogenesi patologica References: 1. Hanahan D, Weinberg RA. Cell. 2000;100: Ferrara N. Endocr Rev ;25: Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23: Ferrara N, Hillan KJ, Gerber HP, Novotny W. Nat Rev Drug Discov. 2004;3: Bergers G, Benjamin LE. Nat Rev Cancer. 2003;3:
, there exists a remarkable similarity in the pathologic traits that collectively drive tumor growth. Across most—if not all—malignancies, sustained angiogenesis is considered to be one of these central cancer hallmarks. 1 The image shown here depicts pathologic angiogenesis. To grow beyond 1 to 2 mm in diameter, a tumor needs an independent blood supply, which is acquired by expressing growth factors that recruit new vasculature from existing blood vessels. This process continues even as the tumor matures. Angiogenesis has been correlated with disease progression and/or poor prognosis in many tumor types—including lung, colon, breast, renal, and other cancers—and can be activated at different stages of tumor development, depending on the tumor type and microenvironmental conditions.2-5. Vasi sanguigni. Angiogenesi patologica. References: 1. Hanahan D, Weinberg RA. Cell. 2000;100: Ferrara N. Endocr Rev. 2004;25: Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23: Ferrara N, Hillan KJ, Gerber HP, Novotny W. Nat Rev Drug Discov. 2004;3: Bergers G, Benjamin LE. Nat Rev Cancer. 2003;3:")
6
The main steps in the formation of a metastasis
The main steps in the formation of a metastasis. a | Cellular transformation and tumour growth. Growth of neoplastic cells must be progressive, with nutrients for the expanding tumour mass initially supplied by simple diffusion. b | Extensive vascularization must occur if a tumour mass is to exceed 1–2 mm in diameter39. The synthesis and secretion of angiogenic factors establish a capillary network from the surrounding host tissue39. c | Local invasion of the host stroma by some tumour cells occurs by several parallel mechanisms40. Thin-walled venules, such as lymphatic channels, offer very little resistance to penetration by tumour cells and provide the most common route for tumour-cell entry into the circulation12, 41. d | Detachment and embolization of single tumour cells or aggregates occurs next, most circulating tumour cells being rapidly destroyed. After the tumour cells have survived the circulation, they become trapped in the capillary beds of distant organs by adhering either to capillary endothelial cells or to subendothelial basement membrane that might be exposed27. e | Extravasation occurs next — probably by mechanisms similar to those that operate during invasion. f | Proliferation within the organ parenchyma completes the metastatic process. To continue growing, the micrometastasis must develop a vascular network39 and evade destruction by host defences. The cells can then invade blood vessels, enter the circulation and produce additional metastases6, 7.

7
The classical angiogenic switch
The classical angiogenic switch. The angiogenic switch is a discrete step in tumour development that can occur at different stages in the tumour-progression pathway, depending on the nature of the tumour and its microenvironment. Most tumours start growing as avascular nodules (dormant) (a) until they reach a steady-state level of proliferating and apoptosing cells. The initiation of angiogenesis, or the 'angiogenic switch', has to occur to ensure exponential tumour growth. The switch begins with perivascular detachment and vessel dilation (b), followed by angiogenic sprouting (c), new vessel formation and maturation, and the recruitment of perivascular cells (d). Blood-vessel formation will continue as long as the tumour grows, and the blood vessels specifically feed hypoxic and necrotic areas of the tumour to provide it with essential nutrients and oxygen (e).
 (a) until they reach a steady-state level of proliferating and apoptosing cells. The initiation of angiogenesis, or the angiogenic switch , has to occur to ensure exponential tumour growth. The switch begins with perivascular detachment and vessel dilation (b), followed by angiogenic sprouting (c), new vessel formation and maturation, and the recruitment of perivascular cells (d). Blood-vessel formation will continue as long as the tumour grows, and the blood vessels specifically feed hypoxic and necrotic areas of the tumour to provide it with essential nutrients and oxygen (e).")
8
Figure 13.34b The Biology of Cancer (© Garland Science 2007)
")
9
Simplified overview of some key steps in tumour angiogenesis
Simplified overview of some key steps in tumour angiogenesis. Tumour cells release pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), which diffuse into nearby tissues and bind to receptors on the endothelial cells of pre-existing blood vessels, leading to their activation. Such interactions between endothelial cells and tumour cells lead to the secretion and activation of various proteolytic enzymes, such as matrix metalloproteinases (MMPs), which degrade the basement membrane and the extracellular matrix. Degradation allows activated endothelial cells — which are stimulated to proliferate by growth factors — to migrate towards the tumour. Integrin molecules, such as v 3-integrin, help to pull the sprouting new blood vessel forward. The endothelial cells deposit a new basement membrane and secrete growth factors, such as platelet-derived growth factor (PDGF), which attract supporting cells to stabilize the new vessel. PDGFR, PDGF receptor; VEGFR, VEGF receptor.
, which diffuse into nearby tissues and bind to receptors on the endothelial cells of pre-existing blood vessels, leading to their activation. Such interactions between endothelial cells and tumour cells lead to the secretion and activation of various proteolytic enzymes, such as matrix metalloproteinases (MMPs), which degrade the basement membrane and the extracellular matrix. Degradation allows activated endothelial cells — which are stimulated to proliferate by growth factors — to migrate towards the tumour. Integrin molecules, such as v 3-integrin, help to pull the sprouting new blood vessel forward. The endothelial cells deposit a new basement membrane and secrete growth factors, such as platelet-derived growth factor (PDGF), which attract supporting cells to stabilize the new vessel. PDGFR, PDGF receptor; VEGFR, VEGF receptor.")
10
The angiogenic balance
The angiogenic balance. Angiogenesis is orchestrated by a variety of activators and inhibitors — only a few of which are listed above. Activators of endothelial-cell proliferation and migration are mainly receptor tyrosine kinase ligands12, such as vascular endothelial growth factor (VEGF), fibroblast growth factors (FGFs), platelet-derived growth factor (PDGF) and epidermal growth factor (EGF), but can also be of very different origin, such as lysophosphatic acid (LPA)89. EGF upregulates VEGF, FGF and interleukin-8, whereas LPA upregulates VEGF levels. The first described angiogenic inhibitor was thrombospondin-1, which modulates endothelial-cell proliferation and motility90. Remarkably, many inhibitory molecules, such as 'statins', are derived from larger proteins that have no effect on angiogenesis. Among those that are listed are angiostatin36 (a fragment of plasminogen that binds ATP synthase and annexin II), as well as endostatin37, tumstatin91 and canstatin92 (fragments of collagens that bind to integrins) (see the review article by R. Kalluri on page 420 of this issue). In general, the levels of activators and inhibitors dictate whether an endothelial cell will be in a quiescent or an angiogenic state. It is believed that changes in the angiogenic balance mediate the angiogenic switch.
, fibroblast growth factors (FGFs), platelet-derived growth factor (PDGF) and epidermal growth factor (EGF), but can also be of very different origin, such as lysophosphatic acid (LPA)89. EGF upregulates VEGF, FGF and interleukin-8, whereas LPA upregulates VEGF levels. The first described angiogenic inhibitor was thrombospondin-1, which modulates endothelial-cell proliferation and motility90. Remarkably, many inhibitory molecules, such as statins , are derived from larger proteins that have no effect on angiogenesis. Among those that are listed are angiostatin36 (a fragment of plasminogen that binds ATP synthase and annexin II), as well as endostatin37, tumstatin91 and canstatin92 (fragments of collagens that bind to integrins) (see the review article by R. Kalluri on page 420 of this issue). In general, the levels of activators and inhibitors dictate whether an endothelial cell will be in a quiescent or an angiogenic state. It is believed that changes in the angiogenic balance mediate the angiogenic switch.")
11
IL VEGF È IL PRINCIPALE FATTORE CHE REGOLA L’ANGIOGENESI
Facilita la soparvvivenza dei vasi esistenti Contribuisce alle anomalie vascolari che possono impedire l’accesso dei chemioterapici al tumore Stimola la crescita di nuovi vasi, contribuendo alla progressione tumorale

12
Il sistema del VEGF Ligands Recettori Chinasi 1 VEGF-E VEGF-A VEGF-B 2
PlGF VEGF-C/D ANGIOGENESI Recettori 3 VEGFR-1 Although there are multiple variants of both the VEGF ligand and its receptor, the angiogenic effects of this pathway are primarily mediated through the interaction of VEGF-A (the most common variant, often referred to simply as VEGF) with VEGFR-2. Other factors are thought to play a secondary role in angiogenesis, though many of these factors may also impact additional nonangiogenic pathways.1-3 Chinasi VEGFR-3 VEGFR-2 Il sistema del VEGF References: 1. Ferrara N. Endocr Rev. 2004;25: Hicklin DJ, Ellis LM. J Clin Oncol ;23: Baka S, Clamp AR, Jayson GC. Expert Opin Ther Targets. 2006;10:
 with VEGFR-2. Other factors are thought to play a secondary role in angiogenesis, though many of these factors may also impact additional nonangiogenic pathways.1-3. Chinasi. VEGFR-3. VEGFR-2. Il sistema del VEGF. References: 1. Ferrara N. Endocr Rev. 2004;25: Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23: Baka S, Clamp AR, Jayson GC. Expert Opin Ther Targets. 2006;10:")
13
STABILITÀ GENETICA DEI BERSAGLI ANTITUMORALI
Fattori di crescita autocrini (p.e. PDGF) Fattori di crescita paracrini (p.e. VEGF) Tumore Tumore Vaso sanguigno Vaso sanguigno As observed in preclinical models, considerable variation may exist in the genetic stability of important antitumor targets. For example, kinase receptors on tumor cells may be susceptible to genetic mutation.1 Moreover, the process of tumor proliferation is mediated largely through autocrine signaling, in which a tumor cell secretes an agent, such as platelet-derived growth factor (PDGF), that acts upon the same cell type.2 Genetic mutations within these cells may destabilize the pathway, potentially limiting the ability to continually target this process.3,4 ANGIOGENESI via di trasduzione del segnale paracrina diretta PROLIFERAZIONE TUMORALE mediata principalmente attraverso vie autocrine di trasduzione del segnale I recettori presenti sulle cellule tumorali sono suscettibili a mutazioni L’instabilità delle cellule tumorali limita la possibilità di inbire in modo continuativo una via autocrina di trasduzione del segnale IL VEGF è una proteina geneticamente stabile La relativa stabilità di ligando e recettori fa sì che l’inibizione continuata del sistema del VEGF sia una strategia antitumorale praticabile References: 1. Baselga J. Science. 2006;312: Goustin AS, Leof EB, Shipley GD, Moses HL. Cancer Res. 1986;46: Busse D, Yakes FM, Lenferink AEG, Arteaga CL. Semin Oncol. 2001;28(suppl 16): Bianco R, Troiani T, Tortora G, Ciardiello F. Endocr Relat Cancer. 2005;12(suppl 1):S159-S171.
 Fattori di crescita paracrini (p.e. VEGF) Tumore. Tumore. Vaso sanguigno. Vaso sanguigno. As observed in preclinical models, considerable variation may exist in the genetic stability of important antitumor targets. For example, kinase receptors on tumor cells may be susceptible to genetic mutation.1 Moreover, the process of tumor proliferation is mediated largely through autocrine signaling, in which a tumor cell secretes an agent, such as platelet-derived growth factor (PDGF), that acts upon the same cell type.2 Genetic mutations within these cells may destabilize the pathway, potentially limiting the ability to continually target this process.3,4. ANGIOGENESI via di trasduzione del segnale paracrina diretta. PROLIFERAZIONE TUMORALE mediata principalmente attraverso vie autocrine di trasduzione del segnale. I recettori presenti sulle cellule tumorali sono suscettibili a mutazioni. L’instabilità delle cellule tumorali limita la possibilità di inbire in modo continuativo una via autocrina di trasduzione del segnale. IL VEGF è una proteina geneticamente stabile. La relativa stabilità di ligando e recettori fa sì che l’inibizione continuata del sistema del VEGF sia una strategia antitumorale praticabile. References: 1. Baselga J. Science. 2006;312: Goustin AS, Leof EB, Shipley GD, Moses HL. Cancer Res. 1986;46: Busse D, Yakes FM, Lenferink AEG, Arteaga CL. Semin Oncol. 2001;28(suppl 16): Bianco R, Troiani T, Tortora G, Ciardiello F. Endocr Relat Cancer. 2005;12(suppl 1):S159-S171.")
14
VANTAGGI DELLE TERAPIE ANTI-ANGIOGENICHE
facile accessibilità del bersaglio selettività minore probabilità di sviluppo di fenotipi resistenti

15
L’ESPRESSIONE DEL VEGF È CONTINUA
Espressione di fattori angiogenici durante il ciclo vitale di un tumore VEGF bFGF TGF-1 PIGF PD-ECGF Pleiotrofina VEGF bFGF TGF-1 VEGF bFGF TGF-1 PIGF PD-ECGF VEGF VEGF bFGF TGF-1 PIGF In addition to being genetically stable over time, VEGF is also continuously expressed throughout the development of many tumor types. In fact, VEGF is the only angiogenic factor known to be present throughout the entire tumor life cycle. As the tumor develops, it may begin to activate secondary angiogenic pathways, such as basic fibroblast growth factor (bFGF), transforming growth factor beta (TGFβ), placental growth factor (PlGF), and platelet-derived endothelial cell growth factor (PD-ECGF). As these secondary pathways emerge, VEGF continues to be overexpressed and remains one of the critical mediators of angiogenesis.1-5 L’ESPRESSIONE DEL VEGF È CONTINUA References: 1. Folkman J. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds. Cancer: Principles & Practice of Oncology. Vol 2. 7th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2005: Bergers G, Benjamin LE. Nat Rev Cancer. 2003;3: Jain RK, Duda DG, Clark JW, Loeffler JS. Nat Clin Pract Oncol. 2006;3: Bergers G, Brekken R, McMahon G, et al. Nat Cell Biol. 2000;2: Inoue M, Hagar JH, Ferrara N, et al. Cancer Cell. 2002;1:
, transforming growth factor beta (TGFβ), placental growth factor (PlGF), and platelet-derived endothelial cell growth factor (PD-ECGF). As these secondary pathways emerge, VEGF continues to be overexpressed and remains one of the critical mediators of angiogenesis.1-5. L’ESPRESSIONE DEL VEGF È CONTINUA. References: 1. Folkman J. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds. Cancer: Principles & Practice of Oncology. Vol 2. 7th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2005: Bergers G, Benjamin LE. Nat Rev Cancer. 2003;3: Jain RK, Duda DG, Clark JW, Loeffler JS. Nat Clin Pract Oncol. 2006;3: Bergers G, Brekken R, McMahon G, et al. Nat Cell Biol. 2000;2: Inoue M, Hagar JH, Ferrara N, et al. Cancer Cell. 2002;1:")
16
Anticorpi contro il ligando TKI a basso peso molecolare
STRATEGIE PER L’INIBIZIONE DEL SISTEMA DEL VEGF VEGF 1 Anticorpi contro il ligando VEGFR-1 (flt-1) VEGFR-2 (KDE/flk-1) While VEGF is the predominant mediator of angiogenesis, there are different strategies for inhibiting its pathway. The 2 primary strategies include inhibiting either the VEGF ligand (ie, ligand antibodies or soluble receptors) or the VEGF receptor (eg, tyrosine kinase inhibitors [TKIs] or receptor antibodies).1 Anti-VEGF strategies that specifically target the ligand, such as VEGF antibodies, inhibit only the VEGF pathway, and therefore may inhibit angiogenesis without disrupting other “off target” pathways. Anti-VEGF strategies that target the receptors, such as TKIs, have a wider range of inhibitory effects and may disrupt other secondary pathways that are also mediated through receptor kinases.2-6 In addition, directly targeting the VEGF ligand results in important antivascular effects that may be sustainable. In both preclinical and clinical models, an anti-VEGF antibody significantly reduced microvascular density, intratumoral pressure, and neovascularization.7-10 These effects have been observed to occur rapidly (in some cases, after a single infusion). However, it should also be noted that monoclonal antibodies allow for a favorable pharmacokinetic profile with a relatively long half-life.4 Cellula endoteliale TKI a basso peso molecolare VEGFR-3 (fls-4) 2 References: 1. Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23: Baka S, Clamp AR, Jayson GC. Expert Opin Ther Targets. 2006;10: Presta LG, Chen H, O’Connor SJ, et al. Cancer Res ;57: Jain RK, Duda DG, Clark JW, Loeffler JS. Nat Clin Pract Oncol. 2006;3: Morabito A, De Maio E, Di Maio M, et al. Oncologist. 2006;11: Kerbel RS. Science. 2006;312: Willett CG, Boucher Y, di Tomaso E, et al. Nat Med. 2004;10: Borgström P, Hillan KJ, Sriramarao P, Ferrara N. Cancer Res. 1996;56: Borgström P, Bourdon MA, Hillan KJ, et al. Prostate. 1998;35: Yuan F, Chen Y, Dellian M, et al. Proc Natl Acad Sci USA. 1996;93:
 VEGFR-2 (KDE/flk-1) While VEGF is the predominant mediator of angiogenesis, there are different strategies for inhibiting its pathway. The 2 primary strategies include inhibiting either the VEGF ligand (ie, ligand antibodies or soluble receptors) or the VEGF receptor (eg, tyrosine kinase inhibitors [TKIs] or receptor antibodies).1 Anti-VEGF strategies that specifically target the ligand, such as VEGF antibodies, inhibit only the VEGF pathway, and therefore may inhibit angiogenesis without disrupting other off target pathways. Anti-VEGF strategies that target the receptors, such as TKIs, have a wider range of inhibitory effects and may disrupt other secondary pathways that are also mediated through receptor kinases.2-6. In addition, directly targeting the VEGF ligand results in important antivascular effects that may be sustainable. In both preclinical and clinical models, an anti-VEGF antibody significantly reduced microvascular density, intratumoral pressure, and neovascularization.7-10 These effects have been observed to occur rapidly (in some cases, after a single infusion). However, it should also be noted that monoclonal antibodies allow for a favorable pharmacokinetic profile with a relatively long half-life.4. Cellula endoteliale. TKI a basso peso molecolare. VEGFR-3 (fls-4) 2. References: 1. Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23: Baka S, Clamp AR, Jayson GC. Expert Opin Ther Targets. 2006;10: Presta LG, Chen H, O’Connor SJ, et al. Cancer Res. 1997;57: Jain RK, Duda DG, Clark JW, Loeffler JS. Nat Clin Pract Oncol. 2006;3: Morabito A, De Maio E, Di Maio M, et al. Oncologist. 2006;11: Kerbel RS. Science. 2006;312: Willett CG, Boucher Y, di Tomaso E, et al. Nat Med. 2004;10: Borgström P, Hillan KJ, Sriramarao P, Ferrara N. Cancer Res. 1996;56: Borgström P, Bourdon MA, Hillan KJ, et al. Prostate. 1998;35: Yuan F, Chen Y, Dellian M, et al. Proc Natl Acad Sci USA. 1996;93:")
17
EFFETTI DELL’INIBIZIONE CONTINUATA DEL VEGF
Regressione della rete microvascolare1-4 esistente “Normalizzazione” dei vasi maturi sopravvissuti Inibizione della ricrescita vascolare e della neovascolarizzazione 1 2 3 1 2 Based on preclinical models, it has been proposed that anti-VEGF agents exert continuous antivascular effects throughout tumor development. One of the most rapid of these proposed effects is regression of existing tumor vessels. Direct and rapid changes observed with anti-VEGF agents include a significant reduction in microvascular density.1-4 While some existing microvasculature may be regressed, other surviving mature vasculature may become “normalized.”2,4,5 This reversal of structural and functional abnormalities may improve the vasculature’s capacity for drug delivery.6,7 In addition to these more rapid effects, anti-VEGF agents may also result in ongoing inhibition of both new and recurrent tumor vessel growth.3,8,9 It has been proposed that these effects inhibit tumor growth and metastasis and improve the tumor vasculature’s capacity for effective delivery of antitumor compounds.5,6,7,10 3 References: 1. Lee CG, Heijn M, di Tomaso E, et al. Cancer Res. 2000;60: Willett CG, Boucher Y, di Tomaso E, et al. Nat Med. 2004;10: Inai T, Mancuso M, Hashizume H, et al. Am J Pathol. 2004;165: Yuan F, Chen Y, Dellian M, et al. Proc Natl Acad Sci USA ;93: Tong RT, Boucher Y, Kozin SV, et al. Cancer Res. 2004;64: Jain RK. Nat Med. 2001;7: Jain RK. Science. 2005;307: Borgström P, Hillan KJ, Sriramarao P, Ferrara N. Cancer Res. 1996;56: Borgström P, Bourdon MA, Hillan KJ, et al. Prostate. 1998;35: Warren RS, Yuan H, Matli MR, et al. J Clin Invest. 1995;95:

18
Jubb et al. Nature Reviews Cancer advance online publication;
The median survival benefit of 4.4 months does not persist, with an equivalent fraction of each treatment group (placebo plus IFL (irinotecan, 5-fluorouracil and leucovorin) versus bevacizumab plus IFL) showing progression-free survival at 20 months. Jubb et al. Nature Reviews Cancer advance online publication; published online 13 July 2006 | doi: /nrc1946
 versus bevacizumab plus IFL) showing progression-free survival at 20 months. Jubb et al. Nature Reviews Cancer advance online publication; published online 13 July 2006 | doi: /nrc1946.")
19
While continued VEGF inhibition is thought to maintain important anti-angiogenic effects that keep tumor cells from growing and spreading, cessation of VEGF suppression may diminish those effects. In fact, in preclinical models, withdrawal of an anti-VEGF agent has been shown to result in rapid regrowth of tumor vasculature.1-4 This vessel regrowth may be particularly rapid along the basement membrane tracks left by previously regressed vasculature.3,4 Because of these proposed effects, many clinical trials with anti-VEGF agents have been designed to maintain VEGF inhibition, even in instances in which administration of accompanying antitumor compounds may be modified.5-8 The CD31 staining images presented here show significant vascular regression from baseline (A) after 7 days of treatment with an anti-VEGF agent (B). After withdrawal of the agent, vessel regrowth is observed within 2 days (C) and vascularity returns to baseline levels after 7 days (D).1 Note: the anti-VEGF agent used in this model was AG , a VEGF TKI with a terminal half-life of 2 to 5 hours.9 LA SOSPENSIONE DEL TRATTAMENTO anti-VEGF È SEGUITA DA UNA RAPIDA RICRESCITA VASCOLARE References: 1. Mancuso MR, Davis R, Norberg SM, et al. J Clin Invest. 2006;116: Vosseler S, Mirancea N, Bohlen P, et al. Cancer Res. 2005;65: Baluk P, Hashizume H, McDonald DM. Curr Opin Genet Dev. 2005;15: Inai T, Mancuso M, Hashizume H, et al. Am J Pathol ;165: Hurwitz H, Fehrenbacher L, Novotny W, et al. N Engl J Med. 2004;350: Sandler A, Gray R, Perry MC, et al. N Engl J Med. 2006;355: Motzer RJ, Michaelson MD, Redman BG, et al. J Clin Oncol. 2006;24: Ratain MJ, Eisen T, Stadler WM, et al. J Clin Oncol. 2006;24: Rugo HS, Herbst RS, Liu G, et al. J Clin Oncol. 2005;23:
 after 7 days of treatment with an anti-VEGF agent (B). After withdrawal of the agent, vessel regrowth is observed within 2 days (C) and vascularity returns to baseline levels after 7 days (D).1 Note: the anti-VEGF agent used in this model was AG , a VEGF TKI with a terminal half-life of 2 to 5 hours.9. LA SOSPENSIONE DEL TRATTAMENTO anti-VEGF È SEGUITA DA UNA RAPIDA RICRESCITA VASCOLARE. References: 1. Mancuso MR, Davis R, Norberg SM, et al. J Clin Invest. 2006;116: Vosseler S, Mirancea N, Bohlen P, et al. Cancer Res. 2005;65: Baluk P, Hashizume H, McDonald DM. Curr Opin Genet Dev. 2005;15: Inai T, Mancuso M, Hashizume H, et al. Am J Pathol. 2004;165: Hurwitz H, Fehrenbacher L, Novotny W, et al. N Engl J Med. 2004;350: Sandler A, Gray R, Perry MC, et al. N Engl J Med. 2006;355: Motzer RJ, Michaelson MD, Redman BG, et al. J Clin Oncol. 2006;24: Ratain MJ, Eisen T, Stadler WM, et al. J Clin Oncol. 2006;24: Rugo HS, Herbst RS, Liu G, et al. J Clin Oncol. 2005;23:")
20
Progressione attraverso vie non-mutazionali
1 VEGF Tumore bFGF TGF-1 PIGF 2 With agents that target VEGF directly, tumor progression may develop through nonmutational pathways. As we saw earlier, VEGF is a genetically stable protein that is continually expressed throughout tumor development.1,2 As the tumor matures, however, secondary angiogenic pathways may become activated that contribute to further tumor development. Activation of these redundant pathways may occur as a compensatory response to treatment or as a result of tumor cell mutations (ie, mutations that are not associated with direct VEGF inhibition).3-6 Because VEGF remains the predominant mediator of angiogenesis, one area of anti-angiogenesis research that is being pursued is maintaining direct VEGF inhibition as tumors progress and supplementing it with selective targeting of other emergent pathways.3,7 Vaso sanguigno Progressione attraverso vie non-mutazionali References: 1. Folkman J. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds. Cancer: Principles & Practice of Oncology. Vol 2. 7th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2005: Mukhopadhyay D, Datta K. Semin Cancer Biol. 2004;14: Sweeney CJ, Miller KD, Sledge GW. Trends Mol Med. 2003;9: Ton NC, Jayson GC. Curr Pharm Des. 2004;10: Kerbel RS, Yu J, Tran J, et al. Cancer Metastasis Rev. 2001;20: Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23: Herbst RS. Expert Opin Emerging Drugs. 2006;11:
.3-6 Because VEGF remains the predominant mediator of angiogenesis, one area of anti-angiogenesis research that is being pursued is maintaining direct VEGF inhibition as tumors progress and supplementing it with selective targeting of other emergent pathways.3,7. Vaso sanguigno. Progressione attraverso vie non-mutazionali. References: 1. Folkman J. In: DeVita VT Jr, Hellman S, Rosenberg SA, eds. Cancer: Principles & Practice of Oncology. Vol 2. 7th ed. Philadelphia, Pa: Lippincott Williams & Wilkins; 2005: Mukhopadhyay D, Datta K. Semin Cancer Biol. 2004;14: Sweeney CJ, Miller KD, Sledge GW. Trends Mol Med. 2003;9: Ton NC, Jayson GC. Curr Pharm Des. 2004;10: Kerbel RS, Yu J, Tran J, et al. Cancer Metastasis Rev. 2001;20: Hicklin DJ, Ellis LM. J Clin Oncol. 2005;23: Herbst RS. Expert Opin Emerging Drugs. 2006;11:")
22
Genetic mechanisms in human tumours that lead to uncontrolled activation of oncogenic tyrosine kinases. a | Reciprocal chromosomal translocations generate fusion proteins, the amino-terminal portion of which is responsible for oligomerization, allowing constitutive activation of the catalytic activity of a kinase domain that is located on the carboxy-terminal portion. b | Overexpression of a cell-membrane receptor tyrosine kinase leads to spontaneous (or autocrine ligand-dependent) dimerization and constitutive kinase activation. c | Point mutation in the juxtamembrane region of a receptor tyrosine kinase causes constitutive ligand-independent dimerization of the receptor and activation of its kinase activity. d | Truncation of the carboxy-terminal portion prevents phosphorylation of a tyrosine residue that is involved in protein folding and stabilizes tyrosine kinase in the active conformation. ABL, Abelson leukaemia; ALK, anaplastic lymphoma kinase; FGFR, fibroblast growth-factor receptor; KD, kinase domain; PDGFR , platelet-derived growth-factor receptor- ; TRKC, neurotrophin 3 receptor.
 dimerization and constitutive kinase activation. c | Point mutation in the juxtamembrane region of a receptor tyrosine kinase causes constitutive ligand-independent dimerization of the receptor and activation of its kinase activity. d | Truncation of the carboxy-terminal portion prevents phosphorylation of a tyrosine residue that is involved in protein folding and stabilizes tyrosine kinase in the active conformation. ABL, Abelson leukaemia; ALK, anaplastic lymphoma kinase; FGFR, fibroblast growth-factor receptor; KD, kinase domain; PDGFR , platelet-derived growth-factor receptor- ; TRKC, neurotrophin 3 receptor..")
23
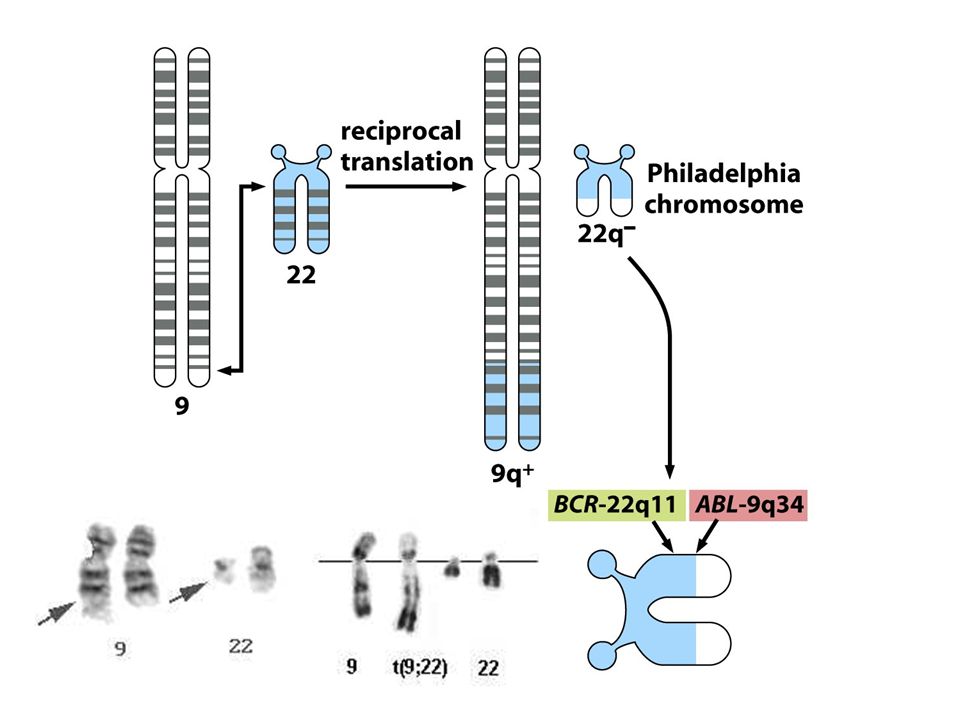
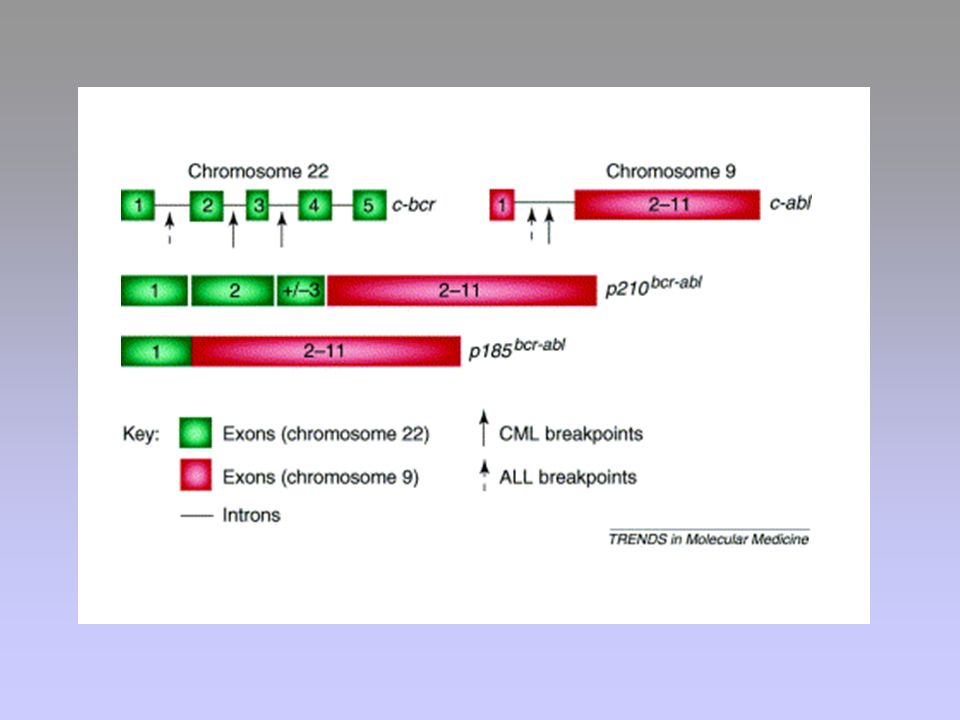
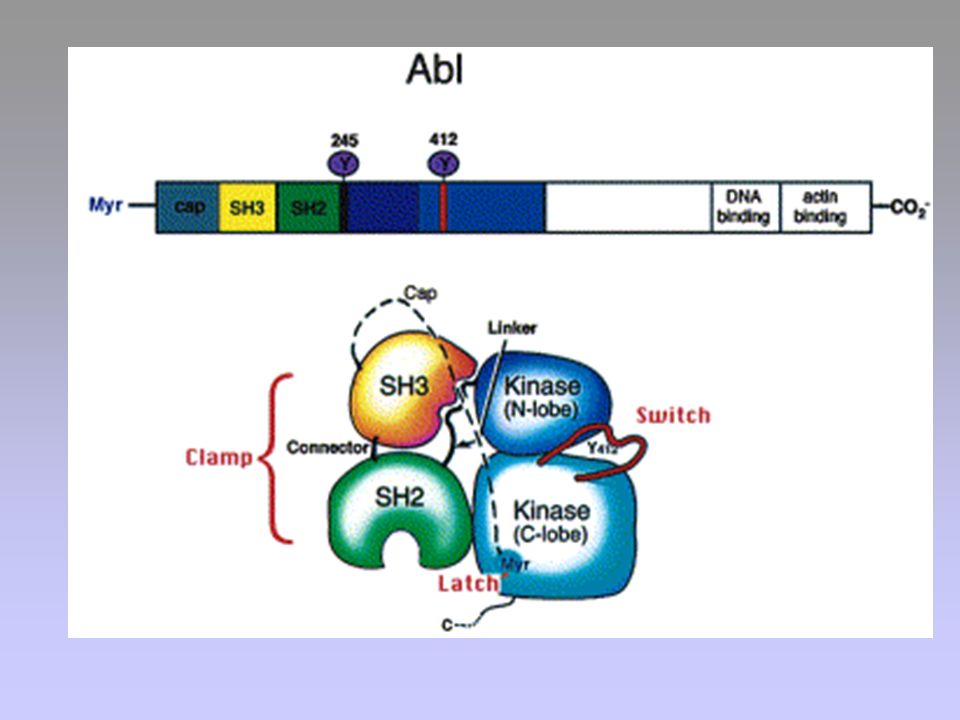
Bcr-Abl

24
LEUCEMIA MIELOIDE CRONICA (CML)
Chronic phase Advanced phase

25
In chronic phase (CP), the bulk of leukaemic stem cells remain capable of undergoing differentiation, leading to the excessive production of mature granulocytes. In advanced phase disease, differentiation has become arrested, probably at the stage of the leukaemia progenitor cell, and the 'aggressive' disease phenotype is caused by the proliferation (self-renewal) of immature blasts. Deleterious genetic events (inset) are believed to accumulate within stem and progenitor cells of the leukaemic clone until there are sufficient secondary mutations to drive the transition from chronic to advanced phase disease. These include: an increase in genomic instability through interference with genomic surveillance and DNA-repair proteins and a progressive telomere shortening. In CP cells essential tumour-suppressor (TS) proteins remain functional and allow cells to undergo replicative senescence or apoptosis. However, in advanced phase blasts there is evidence that TS function has been lost.
 of immature blasts. Deleterious genetic events (inset) are believed to accumulate within stem and progenitor cells of the leukaemic clone until there are sufficient secondary mutations to drive the transition from chronic to advanced phase disease. These include: an increase in genomic instability through interference with genomic surveillance and DNA-repair proteins and a progressive telomere shortening. In CP cells essential tumour-suppressor (TS) proteins remain functional and allow cells to undergo replicative senescence or apoptosis. However, in advanced phase blasts there is evidence that TS function has been lost..")
28
FUNZIONI DI c-Abl Riconoscimento di lesioni a carico del DNA
Progressione del ciclo cellulare Effetti trascrizionali

30
AUMENTO DELLA PROLIFERAZIONE INIBIZIONE DELL’APOPTOSI
BCR SH3 SH2 KINASE DNA BD ACTIN BD Ras, Raf, MAPK PI3K, Akt/PKB STAT5 NFB AUMENTO DELLA PROLIFERAZIONE INIBIZIONE DELL’APOPTOSI

31
(Glivec; Gleevec; Imatinib)
")
32
Figure 1. Imatinib-induced reduction of CML disease burden
Figure 1. Imatinib-induced reduction of CML disease burden. At diagnosis, chronic-phase CML patients have a disease burden of >1012 leukemia cells. Upon imatinib therapy, >95% of newly diagnosed CML patients re-establish normal blood counts, a process termed complete hematologic response (CHR). The curved arrow indicates progressive levels of response among patients achieving CHR. Non-responders to imatinib therapy ( 5%) are indicated in grey. Most patients (>85%) experience at least a three-log reduction in CML disease burden after imatinib therapy, to a level categorized as minimal residual disease (MRD). Failure to reach this level is viewed as a poor prognostic indicator. Disease levels below 109–1010 leukemic cells generally correspond with complete cytogenetic response, defined as the absence of the t(9;22) in either 20 metaphase cells in a bone marrow aspirate or upon sampling of at least 200 cells in a bone marrow aspirate by fluorescence in situ hybridization. Molecular responses are common, but few patients (<5%) reach the level of PCR negativity (complete molecular response). Thus, almost all responding patients have a residual leukemia burden of >106–107 cells. Measurements of disease burden do not reveal which cell types are susceptible to therapy and which are spared. Thomas O’Hare, Amie S Corbin and Brian J Druker Curr Opin Genet Dev Feb;16(1):92-9. Epub 2005 Dec 15
. The curved arrow indicates progressive levels of response among patients achieving CHR. Non-responders to imatinib therapy ( 5%) are indicated in grey. Most patients (>85%) experience at least a three-log reduction in CML disease burden after imatinib therapy, to a level categorized as minimal residual disease (MRD). Failure to reach this level is viewed as a poor prognostic indicator. Disease levels below 109–1010 leukemic cells generally correspond with complete cytogenetic response, defined as the absence of the t(9;22) in either 20 metaphase cells in a bone marrow aspirate or upon sampling of at least 200 cells in a bone marrow aspirate by fluorescence in situ hybridization. Molecular responses are common, but few patients (<5%) reach the level of PCR negativity (complete molecular response). Thus, almost all responding patients have a residual leukemia burden of >106–107 cells. Measurements of disease burden do not reveal which cell types are susceptible to therapy and which are spared. Thomas O’Hare, Amie S Corbin and Brian J Druker Curr Opin Genet Dev Feb;16(1):92-9. Epub 2005 Dec 15.")
33
Figure 1. Schematic representation of the mechanism of action of STI571. The bcr-abl tyrosine kinase is a constitutively active kinase which functions by binding ATP and transferring phosphate from ATP to tyrosine residues on various substrates. This causes the excess proliferation of myeloid cells characteristic of CML. STI571 functions by blocking the binding of ATP to the bcr-abl tyrosine kinase, inhibiting its activity. In the absence of tyrosine kinase activity, substrates required for bcr-abl function cannot be phosphorylated and subsequent cellular events are abrogated.

34
Meccanismi di Attivazione dei recettori ErbB nei Tumori
Attivazione della trasduzione del segnale Mutazione (p.e., EGFRvIII— forma mutante di ErbB1 costitutivamente attiva per delezione della maggior parte del dominio extracellulare) Alterazioni genetiche risultanti nella sovraespressione di recettori normali o costitutivamente attivi Produzione di EGF o TGF-α da parte delle cellule tumorali: creazione di un circuito autocrino che causa l’ attivazione costitutiva di ErbB-1 Activation of ErbB signaling has been associated with a number of different tumor types. There are a variety of different mechanisms that can lead to deregulation, including overexpression of receptors and/or ligands, and genetic mutations producing constitutively active receptors. A number of receptor mutations have been detected in human tumors, but the most common variant is EGFRvIII, a mutated version of ErbB-1.1 This mutation is caused by a deletion of exons 2 through 7, resulting in a truncation of the extracellular domain.1,2 This receptor can no longer bind ligand; however, the mutation results in constitutive activation of the kinase domain and this might contribute to malignant transformation.1 EGFRvIII is not expressed by normal cells, but has been found predominately in gliomas as well as in carcinomas of the prostate, breast, ovary, and NSCLCs.3-6 Dysregulation can also occur through overexpression of the receptor and/or the ligand. In vitro studies suggest that in the case of ErbB-1, expression of the ligand is necessary for transformation by the overexpression of the normal receptor.2 Coexpression of ErbB-1 and its ligands, particularly EGF and TGF-α, is frequently observed in primary tumors, setting up a growth-promoting autocrine signaling loop.2 In contrast, overexpression of the normal ErbB-2 protein alone leads to ligand-independent activation and can thereby alter cellular characteristics such as proliferation, anchorage-independent growth, and tumorigenicity.2 Overexpression of ErbB-2 has been noted in a variety of tumor types, particularly cancers of the breast and ovary.7 Moscatello DK, Holgado-Madruga M, Emlet DR, et al. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem 1998; 273:200-6. 2. Rowinsky E. The erbB receptor family as a target for therapeutic development. Horiz Cancer Ther 2001; 2:3-35. 3. Wikstrand CJ, McLendon RE, Friedman AH, et al. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 1997; 57: 4. Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res 1995; 55: 5. Moscatello DK, Montgomery RB, Sundareshan P, et al. Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene 1996; 13:85-96. 6. Olapade-Olaopa EO, Moscatello DK, MacKay EH, et al. Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer 2000; 82: 7. Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244: Circuito autocrino
 Alterazioni genetiche risultanti nella sovraespressione di recettori normali o costitutivamente attivi. Produzione di EGF o TGF-α da parte delle cellule tumorali: creazione di un circuito autocrino che causa l’ attivazione costitutiva di ErbB-1. Activation of ErbB signaling has been associated with a number of different tumor types. There are a variety of different mechanisms that can lead to deregulation, including overexpression of receptors and/or ligands, and genetic mutations producing constitutively active receptors. A number of receptor mutations have been detected in human tumors, but the most common variant is EGFRvIII, a mutated version of ErbB-1.1 This mutation is caused by a deletion of exons 2 through 7, resulting in a truncation of the extracellular domain.1,2 This receptor can no longer bind ligand; however, the mutation results in constitutive activation of the kinase domain and this might contribute to malignant transformation.1 EGFRvIII is not expressed by normal cells, but has been found predominately in gliomas as well as in carcinomas of the prostate, breast, ovary, and NSCLCs.3-6. Dysregulation can also occur through overexpression of the receptor and/or the ligand. In vitro studies suggest that in the case of ErbB-1, expression of the ligand is necessary for transformation by the overexpression of the normal receptor.2 Coexpression of ErbB-1 and its ligands, particularly EGF and TGF-α, is frequently observed in primary tumors, setting up a growth-promoting autocrine signaling loop.2. In contrast, overexpression of the normal ErbB-2 protein alone leads to ligand-independent activation and can thereby alter cellular characteristics such as proliferation, anchorage-independent growth, and tumorigenicity.2 Overexpression of ErbB-2 has been noted in a variety of tumor types, particularly cancers of the breast and ovary.7. Moscatello DK, Holgado-Madruga M, Emlet DR, et al. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem 1998; 273: Rowinsky E. The erbB receptor family as a target for therapeutic development. Horiz Cancer Ther 2001; 2: Wikstrand CJ, McLendon RE, Friedman AH, et al. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 1997; 57: Wikstrand CJ, Hale LP, Batra SK, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res 1995; 55: Moscatello DK, Montgomery RB, Sundareshan P, et al. Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene 1996; 13: Olapade-Olaopa EO, Moscatello DK, MacKay EH, et al. Evidence for the differential expression of a variant EGF receptor protein in human prostate cancer. Br J Cancer 2000; 82: Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244: Circuito autocrino.")
35
Strategie per l’inibizione dei recettori ErbB
MAbs in grado di bloccare l’interazione con il ligando o la dimerizzazione dei recettori Inibitori chinasici a basso peso molecolare Antagonisti competitivi del recettore Coniugati ligando-tossina o Ab-tossina Oligonucleotidi antisenso Vaccini MAb Ligando- tossina Antagonista Inibitore chinasico There are multiple strategies that could potentially be used to block signaling through the ErbB receptors: Monoclonal antibodies (MoAbs) directed toward the extracellular domain of the receptor can be used to prevent interactions with ligands. This approach might also modulate signaling, dimerization, or receptor expression on the cell surface, as well as potentially triggering antibody-dependent cellular cytotoxicity or complement-mediated cytotoxicity. Small molecules directed toward the kinase domain can inhibit phosphorylation and activation of downstream signaling pathways. Receptor antagonists can be used to competitively block ligand binding. Ligands or receptor-specific antibodies can be conjugated to lethal toxins. Following binding to the receptor, the toxin is internalized and kills the tumor cells. Antisense oligonucleotides can be used to downregulate the expression of ErbB receptors or ligands. Vaccines can be made to trigger the immune system to attack tumor cells overexpressing normal or mutant ErbB receptors. While all of these strategies could potentially be used to inhibit ErbB receptors, so far MoAbs and small-molecule kinase inhibitors have been developed to the greatest extent in a clinical setting.
 directed toward the extracellular domain of the receptor can be used to prevent interactions with ligands. This approach might also modulate signaling, dimerization, or receptor expression on the cell surface, as well as potentially triggering antibody-dependent cellular cytotoxicity or complement-mediated cytotoxicity. Small molecules directed toward the kinase domain can inhibit phosphorylation and activation of downstream signaling pathways. Receptor antagonists can be used to competitively block ligand binding. Ligands or receptor-specific antibodies can be conjugated to lethal toxins. Following binding to the receptor, the toxin is internalized and kills the tumor cells. Antisense oligonucleotides can be used to downregulate the expression of ErbB receptors or ligands. Vaccines can be made to trigger the immune system to attack tumor cells overexpressing normal or mutant ErbB receptors. While all of these strategies could potentially be used to inhibit ErbB receptors, so far MoAbs and small-molecule kinase inhibitors have been developed to the greatest extent in a clinical setting.")
36
Framework for developing anticancer drugs with a high therapeutic index. An anticancer drug might have a high therapeutic index because its target is uniquely present in cancer cells (a), or because the requirement for its target is quantitatively or qualitatively different in cancer cells than in normal cells (b and c). This differential requirement might be because of intrinsic differences in the cells (b), such as genetic (red) and epigenetic (blue) differences, or extrinsic differences in the cells (c), such as loss of survival signals provided by normal cell–cell and cell–matrix interactions. Modified with permission from Ref. 2 © (2002) Elsevier Science.
, or because the requirement for its target is quantitatively or qualitatively different in cancer cells than in normal cells (b and c). This differential requirement might be because of intrinsic differences in the cells (b), such as genetic (red) and epigenetic (blue) differences, or extrinsic differences in the cells (c), such as loss of survival signals provided by normal cell–cell and cell–matrix interactions. Modified with permission from Ref. 2 © (2002) Elsevier Science..")
37
Framework for developing anticancer drugs with a high therapeutic index. An anticancer drug might have a high therapeutic index because its target is uniquely present in cancer cells (a), or because the requirement for its target is quantitatively or qualitatively different in cancer cells than in normal cells (b and c). This differential requirement might be because of intrinsic differences in the cells (b), such as genetic (red) and epigenetic (blue) differences, or extrinsic differences in the cells (c), such as loss of survival signals provided by normal cell–cell and cell–matrix interactions. Modified with permission from Ref. 2 © (2002) Elsevier Science.
, or because the requirement for its target is quantitatively or qualitatively different in cancer cells than in normal cells (b and c). This differential requirement might be because of intrinsic differences in the cells (b), such as genetic (red) and epigenetic (blue) differences, or extrinsic differences in the cells (c), such as loss of survival signals provided by normal cell–cell and cell–matrix interactions. Modified with permission from Ref. 2 © (2002) Elsevier Science..")
38
+ Time Death or failure to proliferate

39
Survival signals in tumour cells: an Achille's heel?
The issue of the degree of the therapeutic window that will be provided by drugs that target the RAS pathways is a crucial one. All cells use RAS signalling pathways to some extent, so there is a danger that inhibitors will have severe effects on normal cells as well as tumour cells. Potent inhibition of RAS function through the expression of dominant-negative mutants or microinjection of neutralizing antibodies has long been known to block normal cell proliferation. Although, ultimately, each drug target has to be validated experimentally for its differential effect on tumour versus normal cells, there are conceptual reasons for believing that certain types of signalling inhibitors, in particular those that inhibit survival pathways, might selectively disadvantage tumour cells. As represented in the figure by the 'scales' of life and death signals that the cell is experiencing, a normal cell requires a continuous low level of survival signal to remain alive85. These survival signals emanate from various different sources, including adhesion to extracellular matrix, soluble factors in the extracellular environment and interactions between cells. Each of these acts to instruct the cell that it is in an appropriate environment. The cells are also exposed to a low level of cell-death signals, perhaps due to occasional DNA damage or oxidative stress, but the balance of signals favours survival. In the tumour cells, microevolutionary processes have led to the selection of cells with greatly increased survival signalling — for example, by the loss of PTEN. Once a mutation has given a cell a survival advantage, it is then able to tolerate more death signals. For example, this allows it to survive notable DNA damage as a result of loss of cell-cycle control and rapid proliferation, and to be less easily killed by hypoxia and by immune system attack; all of these events result in the tumour cell's increasingly antisocial behaviour. It can also afford to dispense with previously required survival signals that were provided by matrix and other cells and to grow in a completely independent manner that is characteristic of tumour cells. This disorganized growth is therefore occurring at high levels of both apoptotic and survival signals, with the cells being dependent on one or a few strongly activated survival pathways, compared with a more complex pattern of survival signals for the normal cell. If both cell types are now treated with a survival-pathway inhibitor that targets the pathway on which the tumour cell is reliant, rapid death of the tumour cell will result, driven by the damage it has accumulated and formerly been able to ignore. The normal cell, by contrast, might survive, having much lower intrinsic levels of death signals and receiving a wider range of survival signals.

Presentazioni simili





>")










>")
 to different types of stress (blue boxes). Activation of p53 can result in a number.>")
 Advanced phase.>")

